CHAPTER 16
SALIVARY GLAND LYMPHOMAS
Primary lymphomas of salivary glands are rare and represent less than 2% of primary salivary gland neoplasms. Similar to lymph nodes, most salivary gland lymphomas are derived from mature B cells. Marginal zone lymphoma of mucosa-associated lymphoid tissue is the most common salivary gland lymphoma and frequently develops in association with reactive lymphoid proliferations in autoimmune conditions. Less common are follicular lymphoma and diffuse large B-cell lymphoma. The latter can be primary or develop as a transformation of low-grade lymphoid neoplasm. Parotid and submandibular glands are the predominant sites of involvement. Minor salivary gland lymphomas are less common.
Because of the overlap in cytologic features, the definitive diagnosis and subclassification of salivary gland lymphoma on cytology alone is frequently challenging. Cytologic evaluation of fine needle aspiration (FNA) commonly is accompanied by immunophenotyping by flow cytometry or immunohistochemistry of a cell block.
16.2.1 Introduction
Hodgkin lymphoma, formerly known as Hodgkin disease, has been described first by Thomas Hodgkin in 1832. The controversy over the origin of this lymphoma lasted more than a century and led to the original designation as Hodgkin disease or lymphogranulomatosis. With the demonstration of the B-cell origin of this neoplasm, the term Hodgkin lymphoma has been introduced. The World Health Organization (WHO) classification of lymphoid and hematopoietic tumors divides Hodgkin lymphoma into two categories—nodular lymphocyte-predominant Hodgkin lymphoma and classical Hodgkin lymphoma (cHL). Nodular lymphocyte-predominant Hodgkin lymphoma consists of lymphocytic/histiocytic or “popcorn” cells scattered within nodules of reactive lymphocytes. It rarely involves extranodal sites and thus is not discussed in this chapter in detail. cHL is characterized by the presence of Reed-Sternberg (RS) cells in a rich reactive inflammatory background. The cHL is observed most frequently in lymph nodes and other lymphoid organs such as the spleen and liver. It occasionally presents in extranodal sites, especially in immunodeficient individuals. The cHL is divided into the following histologic subtypes: nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted.
16.2.2 Clinical Features
Classical Hodgkin lymphoma presents most frequently in lymph nodes and can involve related lymphoid organs such as the spleen and liver. Cervical lymphadenoapthy is most common followed by mediastinal, axillary, or paraaortic lymph node involvement. Bone marrow is involved in 5% of cases. Primary extranodal site cHL is rare in immunocompetent individuals and is more frequent in the immunodeficient population. Rare reports of salivary gland involvement have been published in the literature. B symptoms including fever, weight loss, and night sweats are common in cHL.
16.2.3 Cytologic Features with Histologic Correlates
Fine needle aspirations of cHL can be variably cellular depending on the histologic subtype of the lesion. The lymphocyte-rich and mixed cellularity types usually yield a richly cellular sample with most cells being small lymphocytes admixed with rare neoplastic cells. The nodular sclerosis cHL may be difficult to aspirate because of extensive fibrosis. The aspirate can contain fibroblasts and metachromatic fibrillar material derived from collagenous bands characteristic of nodular sclerosis cHL.
Regardless of the histologic subtype, the pathognomonic findings are the presence of RS cells in a reactive inflammatory background comprising small lymphocytes, plasma cells, histiocytes, and eosinophils (Figure 16.1a and b, Table 16.1). Granulomatous reaction and necrotic foci can be present. The RS cells are required for the diagnosis and are present in each histologic subtype of cHL. The typical RS cell is a large lymphoid cell, of an average size of 40–70 μm, with a bilobed nucleus or two nuclei with prominent centrally located inclusion-like nucleolus and abundant cytoplasm (Figure 16.1c). The appearance of the nuclei sometimes is compared with the image of an owl’s eyes. In Papanicolaou stain, the characteristic nucleoli of RS cells stain bright red. The cytoplasm is abundant and stains usually blue-green in Papanicolaou and basophilic in Diff-Quik stain. The variants of RS cells such as mononuclear Hodgkin cells, lacunar cells, and mummified cells are observed frequently; however, they are not sufficient for the diagnosis of cHL (Figure 16.1d). Mononuclear Hodgkin cells are large lymphoid cells with a single nucleus, distinct nucleolus, and abundant cytoplasm. Lacunar cells are appreciated best in the histologic sections (needle biopsies or cell blocks). They may be mononuclear with oval or multilobated nuclei, or multinucleated, and show cytoplasm retraction in formalin fixed tissue. Mummified cells have pyknotic nuclei and condensed cytoplasm. Nucleoli are difficult to appreciate. Because select non-Hodgkin lymphomas closely can resemble cHL cytologically, immunohistochemical stains, described in the following paragraphs, are required for a definitive diagnosis.
FIGURE 16.1. Classical Hodgkin lymphoma. (a) FNA of nodular sclerosis cHL showing cohesive aggregates including small lymphocytes, histiocytes, fibroblasts granulocytes, and scattered RS and Hodgkin cells (Papanicolaou, 500×). (b) Occasionally, aspirates of cHL show numerous RS and Hodgkin cells. In such cases, differential diagnosis includes DLBCL and nonlymphoid neoplasms (Diff-Quik, 600×). (c) In Diff-Quik stain, classic RS cells have blue cytoplasm and distinct inclusion-like nucleoli (1000×). (d) Classical RS cells (center) in a histologic section from cHL (hematoxylin and eosin, 1000×).
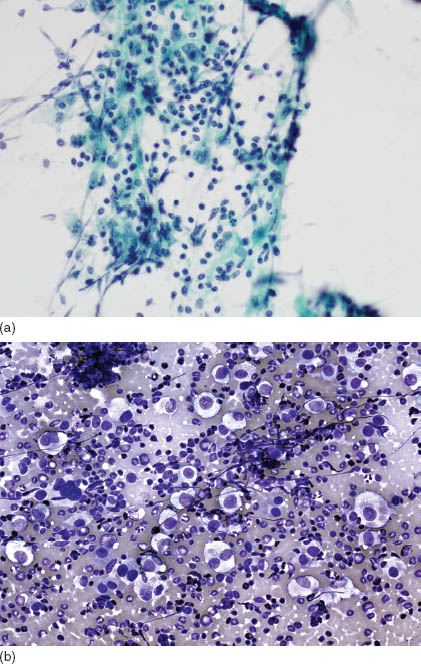
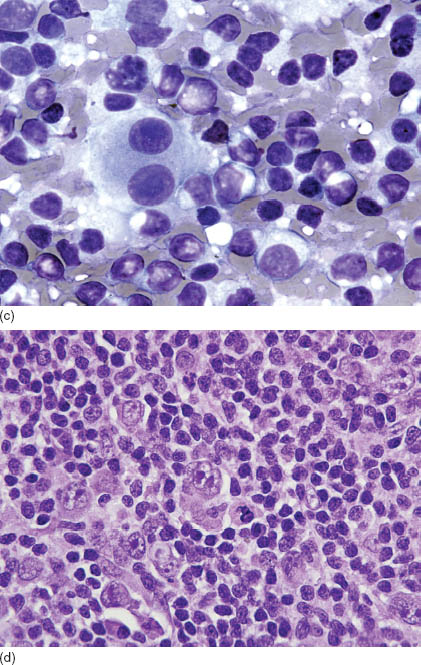
TABLE 16.1. Key diagnostic features of salivary gland lymphomas
| Lymphoma subtype | Key diagnostic features |
| Classical Hodgkin lymphoma | Reed-Sternberg cells in a rich background of small lymphocytes, plasma cells, histiocytes, and eosinophils |
| Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue | Noncohesive polymorphous lymphoid population encompassing predominantly small and medium-sized lymphocytes, with dense chromatin, and oval-to-irregular nuclei. Plasma cells and large lymphoid cells are present |
| Follicular lymphoma | Low-grade (histologic grade 1–2): population of small- to medium-sized centrocytes with cleaved nuclei (can be angular or indented) with noticeable, however, infrequent centroblasts High-grade (histologic grade 3): predominance of centroblasts (large lymphoid cells with oval nuclei and multiple peripheral nucleoli) cytologically indistinguishable from DLBCL |
| Diffuse large B-cell lymphoma | Cellular FNA smear showing numerous noncohesive large lymphoid cells, which can demonstrate nuclear pleomorphism, abundant cytoplasm and prominent nucleoli |
| Plasma cell neoplasm | Noncohesive cells with eccentric nuclei, distinct nucleoli, clock-face chromatin, and perinuclear hof. Intranuclear (Dutcher) and intracytopalsmic (Russel) inclusions may be present. Binucleated forms can be observed |
Histologic features of cHL are dependent on the subtype. Briefly, nodular sclerosis cHL, the most common type accounting for approximately 70% of cHL cases, shows broad collagen bands transsecting the lymph node and thickening of the nodal capsule. Lacunar cells frequently are present in this type of cHL. The background cellularity includes small lymphocytes, eosinophils, and histiocytes. In mixed cellularity cHL, fibrotic bands and capsular thickening are absent, and RS cells are scattered among the diffuse background proliferation of small lymphocytes, histiocytes, eosinophils, neutrophils, and plasma cells. Lymphocyte-rich type shows a vaguely nodular background of small lymphocytes with scattered RS cells. The RS cells and variants are most frequent in lymphocyte-depleted cHL, which is characterized by a paucity of a reactive background.
The demonstration of RS cells positive for CD30 and CD15 and negative for CD45 and T-cell markers is essential for the diagnosis. Immunohistochemical stain for the CD20 antigen can be weakly positive in a proportion of RS cells; however, at the time of primary diagnosis, staining is typically heterogeneous and not as intense as in cases of non-Hodgkin B-cell lymphomas. Relapsed cHL with strong CD20 immunoreactivity has been reported. The cellular composition of background lymphoid cells is variable and dependent on the subtype of cHL. In most cases, CD4-positive T cells predominate. In lymphocyte-rich cHL, RS cells are embedded in remnants of mantle zones and follicles, comprising mainly B-cells; thus, immunophenotyping shows a more prominent B-cell background. Immunophenotype of RS cells is demonstrated best by immunohistochemistry of a cell block.
16.2.4 Differential Diagnosis
Close attention is essential to both the cytology of neoplastic cells and to the reactive background. A host of other lymphoid malignancies and select reactive conditions closely can resemble cHL (Table 16.2). The most common reactive conditions, which cytologically can mimick cHL, are infectious mononucleosis and other reactive lymphoid proliferations containing immunoblasts, which occasionally resemble RS and Hodgkin cells. Reactive immunoblastic proliferations most commonly are associated with viral infections and autoimmune conditions. They typically show a continuum of cell cytology from small lymphocytes, through medium-sized lymphoid cells, and to large transformed lymphocytes. The latter may be bi- or multinucleated and feature large distinct nucleoli. In contrast, in cHL, one usually appreciates isolated RS and Hodgkin cells in an inflammatory background that may include small lymphoid cells, plasma cells, granulocytes, plasma cells, and histiocytes. The intermediate size lymphoid cells and cytologic continuum is not present. Granulomatous foci with epithelioid histiocytes also can be present in cases of cHL; thus, fine needle aspirations of any lesion with granulomatous inflammation have to be scrutinized closely for the presence of RS cells to exclude cHL.
TABLE 16.2 Differential diagnosis of classical Hodgkin lymphoma
| Major differential diagnosis | Clues to make the distinction |
| Reactive conditions containing atypical immunoblasts resembling RS and Hodgkin cells frequently associated with viral infections | Reactive lymphoid proliferations most frequently show a continuum of lymphoid cell cytology from small- to medium-sized to large pleomorphic forms. On the contrary, cHL is characterized by RS cells and variant Hodgkin cells immersed in inflammatory background, no transitional forms, or cytologic continuum is observed |
| T cell/histiocyte-rich large B-cell lymphoma (THRLBCL), and other subtypes of DLBCL | If THRLBCL contain large pleomorphic cells, then it is cytologically indistinguishable from cHL. THRLBCL is rare at extranodal sites, and thus infrequently will be considered in the differential diagnosis. Other DLBCL subtypes usually have frequent large cells, which aid in the differentiation from cHL. In contrast to cHL, all DLBCL are positive for several B-cell markers such as CD20 and CD79a and the panhematopoietic antigen CD45 (LCA) |
| Anaplastic large cell lymphoma | ALCL can present with a rich inflammatory background, and these cases are challenging to distinguish from cHL unless IHC is performed to confirm T-cell/null immunophenotype and/or positivity for the ALK-1 antigen. The presence of numerous neoplastic cells and hallmark cells support the diagnosis of ALCL |
ALCL = anaplastic large cell lymphoma; IHC = immunohistochemistry; LCA = leukocyte common antigen; THRLBCL = T cell/histiocyte-rich large B-cell lymphoma.
The neoplasm most closely resembling cHL on cytologic examination is T cell/histiocyte-rich large B-cell lymphoma (THRLBCL), a subtype of diffuse large B-cell lymphoma (DLBCL). In this entity, rare neoplastic B cells are scattered in the reactive background comprising small lymphocytes with a variable admixture of histiocytes. Malignant cells can be pleomorphic and mimic RS and Hodgkin cells. However, T-cell/histiocyte-rich large B-cell lymphoma rarely involves tissues outside of the lymphoid system or bone marrow and, based on the site of involvement, rarely will enter the differential diagnosis in lesions of salivary glands.
Typical diffuse large B-cell lymphoma more commonly involves salivary glands and in select cases can contain pleomorphic cells resembling RS and Hodgkin cells. In these cases, large cells predominate; thus, the main differential diagnosis will include lymphocyte-depleted or a syncytial variant of nodular sclerosis cHL. In such rare cases, immunohistochemical stains of cell block or flow cytometric immunophenotyping of fine needle aspirate are helpful in rendering a definitive diagnosis. Similarly, an immunophenotype is required for the differentiation between cHL and anaplastic large cell lymphoma, two entities shown to be misdiagnosed most frequently in nodal fine needle aspirates. Anaplastic large cell lymphoma, reported to rarely involve the parotid gland, consists of pleomorphic lymphoid cells with large nuclei, bi- or multinucleation, and abundant cytoplasm. A rich inflammatory background can be present. Wreath-like nuclei of neoplastic cells suggest anaplastic large cell lymphoma; however, final diagnosis requires immunohistochemical stains.
Rarely, other lymphoid malignancies, such as small lymphocytic lymphoma can contain RS-like cells, either at the time of original diagnosis or at the time of transformation (non-Hodgkin lymphoma as Richter transformation). These cases are rare and will require immunophenotyping to assess the nature of the background small lymphocytic proliferation adequately.
Finally, when numerous and clustering of RS and Hodgkin cells are encountered in FNA of a syncytial variant of nodular sclerosis cHL, a differential diagnosis includes poorly differentiated carcinoma, sarcoma, and melanoma.
16.2.5 Treatment and Prognosis
The cHL is treated with combination chemotherapy and/or radiation. The choice of treatment modality is based on the stage of the disease. More than 85% of patients are cured with this approach. Staging and laboratory features are used for prognostication. The best outcome occurs in patients with lymphocyte-rich and nodular sclerosis cHL, and the worst survival has been reported for the lymphocyte-depleted type.
16.3 EXTRANODAL MARGINAL ZONE LYMPHOMA OF MUCOSA-ASSOCIATED LYMPHOID TISSUE (MALT LYMPHOMA)
16.3.1 Introduction
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) is the most common type of primary lymphoma in salivary glands. The original description of Mikulicz disease by Jan Mikulicz-Radecki in 1892 most likely represents the first description of salivary gland MALT lymphoma.
MALT lymphoma develops from postgerminal center memory B-cells, most frequently in patients with an autoimmune disease such as Sjogren syndrome, and is preceded by an acquisition of prominent mucosa-associated lymphoid tissue. The neoplastic population of MALT lymphoma is heterogeneous and includes small- and medium-sized lymphocytes, plasma cells, and occasional large lymphoid cells. The cytologic features overlap to a large extent with myoepithelial/lymphoepithelial sialadenitis (benign lymphoepithelial lesion), which makes cytologic diagnosis of MALT lymphoma one of the most challenging in salivary gland pathology.
16.3.2 Clinical Features
MALT lymphoma of salivary glands is the most frequent subtype of malignant lymphoma developing at this site. Parotid glands predominantly are involved. MALT lymphoma frequently presents with diffuse symmetric bilateral enlargement of salivary glands. Adults are affected most commonly, and there is a significant female predominance. Most patients have a prior history of autoimmune disease, predominantly Sjogren syndrome. This patient group have a 44-fold increased risk of developing lymphoma, which can be preceded by a benign lymphoepithelial lesion. The clinical course is usually indolent, and lymphoma remains confined to salivary glands. Rarely, patients develop disseminated disease and/or transformation to diffuse large B-cell lymphoma. Paraproteinemia may be present.
16.3.3 Cytologic Features with Histologic Correlates
MALT lymphoma consists of a population of small-to-intermediate-sized lymphocytes with abundant cytoplasm, irregular nuclear borders, and occasional nucleoli (monocytoid and centrocyte-like cells; Figure />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


