Rheumatology and orthopaedics
Key Points
• Osteoarthritis is very common, especially in the hips, knees and spine, and with age
• Rheumatoid arthritis is an incapacitating, fairly common systemic condition
Bone
Bone morphogenetic proteins (BMPs) affect bone formation and are available as recombinant proteins, sometimes used in repair of periodontal defects, other bone defects and in implantology. Bone organic matrix consists largely of collagen produced by osteoblasts rich in alkaline phosphatase. Osteoblasts secrete osteoid, which mineralizes by deposition of calcium phosphate, largely as hydroxyapatite, together with smaller amounts of amorphous calcium phosphate, magnesium, sodium and other ions. Mineralization depends on the extracellular fluid calcium and phosphate levels.
Bone is a dynamic tissue, constantly remodelling, with deposition mediated by osteoblasts, and resorption mediated mainly by osteoclasts (also by prostaglandins and some cytokines). When osteoblastic activity rises, the serum level of alkaline phosphatase rises. When bone is resorbed, calcium and phosphate are removed and released into the extracellular fluid and serum. Osteoclasts are rich in acid phosphatase (particularly tartrate-resistant acid phosphatase), which may also be released into the serum when there is widespread osteolysis. Bone formation, metabolism and blood calcium levels are affected mainly by parathyroid hormone, vitamin D and calcitonin. Parathyroid hormone (PTH; parathormone) is regulated by blood calcium, vitamin D levels and phosphate levels. When PTH is secreted, intestinal transport of calcium and phosphate is promoted, and removal of calcium from bones accelerated.
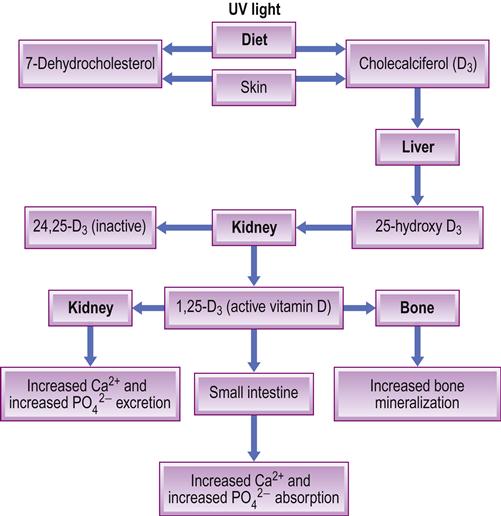
Dietary vitamin D is fat-soluble and absorbed from the upper small intestine, promoting intestinal absorption of calcium and phosphate. Vitamin D is also synthesized in the skin from 7-dehydrocholesterol under the influence of sunlight, and converted by liver and then kidney to the most active metabolite, 1,25-dihydroxycholecalciferol (DHCC: calcitriol – the hormonal form of vitamin D), a process enhanced by PTH and low phosphate levels. The active metabolite (active vitamin D or DHCC) is a hormone that controls bone metabolism and enhances calcium absorption (Fig. 16.1). DHCC also, incidentally, affects the proliferation of various cells other than bone and enhances immunity. Vitamin D exerts its influence on many physiological processes, including the immune system; both the adaptive and innate immune systems are impacted by the active metabolite of vitamin D, 1,25(OH)(2)D, and individuals with vitamin D deficiency are prone to infectious diseases such as tuberculosis, as well as to autoimmune diseases such as type 1 diabetes mellitus, and to multiple sclerosis. Calcitonin opposes the action of PTH and lowers the blood calcium level, mainly by promoting deposition of calcium in the bones.
Bone growth varies throughout life; it is rapid in the fetus and infant, but slows somewhat during childhood until an adolescent growth spurt. Several other hormones affect bone formation and metabolism to varying degrees, especially during the first two decades of life, including growth hormone, oestrogens in females and testosterone in males. Adult levels of bone mass are achieved by age 18 or so.
Lifestyle, especially physical activity and adequate nutrition, promotes the formation of bone, whereas smoking may impair it. Osteoblasts are also less efficient at laying down bone than are osteoclasts at removing it and, although the difference is slight, there is a gradual loss of bone mineral density (BMD) as a person ages (osteopenia). Anything that accelerates the rate of bone remodelling ultimately leads to a more rapid loss of bone mass and thus more fragile bones (osteoporosis).
Bone Diseases
Acquired disorders of the skeleton are common. Genetic disorders are rare; the more important examples are discussed here and main features of others summarized in Appendix 16.1.
Genetic Disorders
Osteogenesis imperfecta (fragilitas ossium; brittle bone syndrome)
General aspects
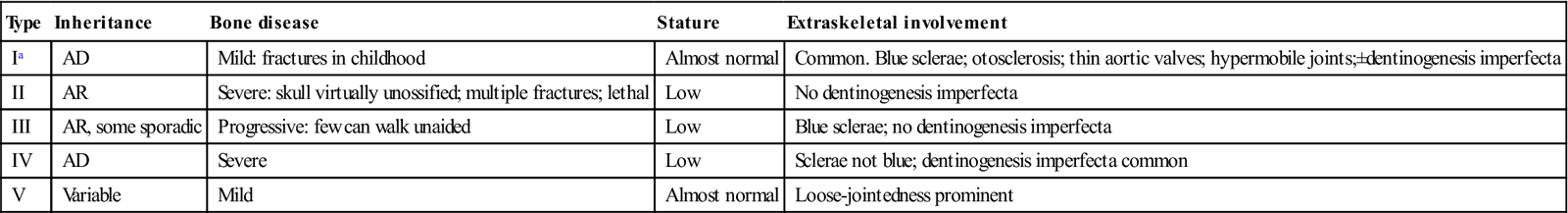
Osteogenesis imperfect (OI) is a rare, usually autosomal dominant, condition characterized by brittle bones that are susceptible to fracture. The underlying defect appears to be in type I collagen formation and, although osteoblasts are active, the amount of bone formed is small and mostly woven in type. This disorder, with a frequency of 1 in 12 000 births, has several variants (Table 16.1). The gene for dentinogenesis imperfecta, one of the more common heritable defects of the teeth, is closely related – and some patients (OI types I and IV) have both conditions.
Table 16.1
Osteogenesis imperfecta: subtypes
< ?comst?>
| Type | Inheritance | Bone disease | Stature | Extraskeletal involvement |
| Ia | AD | Mild: fractures in childhood | Almost normal | Common. Blue sclerae; otosclerosis; thin aortic valves; hypermobile joints;±dentinogenesis imperfecta |
| II | AR | Severe: skull virtually unossified; multiple fractures; lethal | Low | No dentinogenesis imperfecta |
| III | AR, some sporadic | Progressive: few can walk unaided | Low | Blue sclerae; no dentinogenesis imperfecta |
| IV | AD | Severe | Low | Sclerae not blue; dentinogenesis imperfecta common |
| V | Variable | Mild | Almost normal | Loose-jointedness prominent |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Clinical features
The long bones are normal in length, with epiphyses of normal width but slender shafts, giving a trumpet-shaped appearance. The bones are fragile and multiple fractures can follow minimal trauma; healing is rapid but usually with distortion, so the ultimate effect in severe cases is gross deformity and dwarfism. The frequency of fractures usually tends to diminish after puberty.
The parietal regions of the skull may bulge outwards, causing eversion of the upper part of the ear. Other features are blue sclerae, deafness, easy bruising, and weakness of tendons and ligaments, causing loose-jointedness and often hernias. Cardiac complications, such as mitral valve prolapse or aortic incompetence, may be present.
Dental aspects
It is important not to confuse children with OI with those who have been subjected to physical abuse (Ch. 24). The management problems relate to bone fragility and careful handling of the patient. Despite the brittleness of most of the skeleton, however, the jaws rarely fracture and therefore dental extractions can usually be carried out safely; care should obviously be taken to use minimal force, to support the jaws and to ensure haemostasis if there is any bleeding tendency (Ch. 8). GA is a risk if there are chest deformities or cardiac complications.
When dentinogenesis imperfecta is associated, the teeth may have the characteristic abnormal translucency, brown or purplish colour and tendency to wear readily. The enamel may adhere poorly to the dentine and be progressively shed under the stress of mastication. By adolescence, the teeth may be worn down to the gingival margins but obliteration of the pulp chamber by dentine usually prevents pulp exposure. The soft dentine makes the fitting of post crowns impractical so that steel crowns on primary molars and crowns in the permanent dentition, or even extraction and replacement by dentures, may be needed in severe cases. Bisphosphonate treatment may have implications (see Osteonecrosis of the jaw)
Dentinogenesis imperfecta can appear as an isolated abnormality without OI.
Achondroplasia
The achondrodysplasias are heritable disorders of skeletal growth, presenting typically with dwarfism, high forehead, cleft palate and various ocular anomalies.
General and clinical aspects
There are defects in cartilaginous bone formation, often inherited as an autosomal dominant trait. The phenotypic appearance is of dwarfism with a head of normal size but appearing large, and short limbs, which has traditionally led to many achondroplastics becoming circus clowns. Nasal septal and base of skull growth is impaired so that the bridge is depressed and skull bossed. Lumbar lordosis can be severe and occasionally causes spinal cord compression. Many patients with achondroplasia are otherwise completely healthy but diabetes is occasionally associated.
Dental aspects
The only significant dental aspects are orthodontic in nature as a result of malocclusion, but short stature, diabetes and kyphosis may affect management. Surgery is usually required for the cleft palate and other deformities.
Cleidocranial dysplasia (cleidocranial dysostosis)
General aspects
Cleidocranial dysplasia is a rare defect, mainly of membrane bone formation, involving especially the skull and clavicle. It is an autosomal dominant trait related to chromosome 6 and a defect in the signal transduction SH3-binding protein. There are also sporadic cases.
Clinical features
The clavicles are either absent or defective, thus conferring the unusual and pathognomonic ability to approximate the shoulders anteriorly (Fig. 16.2). The head is large but brachycephalic, with persistent fontanelles, a persistent metopic (frontal) suture, numerous Wormian bones and bulging frontal, parietal and occipital bones. The middle facial third is hypoplastic, leading to a depressed nasal bridge and relative mandibular protrusion. The mandibular symphysis may close late. Cranial base abnormalities and clefts of the hard and soft palate have been recorded. Other skeletal defects may be associated, such as kyphoscoliosis or pelvic anomalies.
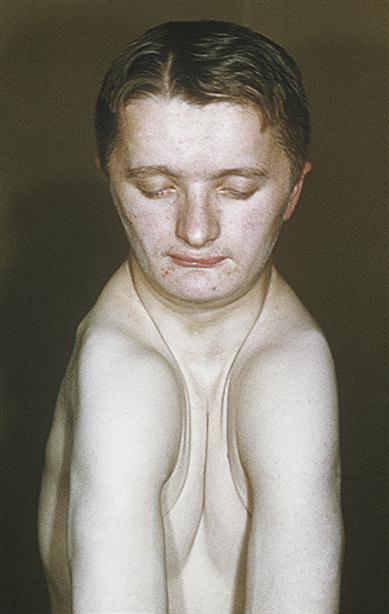
Dental aspects
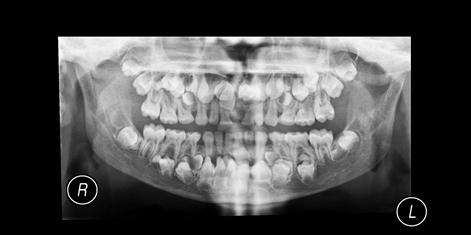
Apart from the facial anomalies, there may be excess teeth (hyperdontia, supernumeraries; Fig. 16.3) with persistence of the deciduous dentition, multiple unerupted permanent teeth, twisted roots, malformed crowns and dentigerous cysts.
Osteopetrosis (Albers–Schönberg disease; marble-bone disease)
General aspects
Osteopetrosis is a rare genetic disorder of variable severity characterized by excessive bone density, probably as a result of a defect in osteoclastic activity and hence of bone remodelling. The bones, though dense, are weak and can fracture easily, but usually heal normally.
Clinical features
In mild osteopetrosis, there may be no symptoms and the diagnosis is made by chance by radiography. In severe osteopetrosis (malignant infantile osteopetrosis), patients may suffer bone pain, fractures or osteomyelitis; added complications can be cranial neuropathies leading, for example, to optic atrophy. Anaemia is common, as the medullary cavities are filled with bone, despite extramedullary haemopoiesis in liver, spleen and lymph nodes, and there is a susceptibility to infections, since macrophages and neutrophils are defective. Hydrocephalus, epilepsy and learning disability are less frequent.
General management
Serum levels of acid phosphatase are raised but calcium and phosphate levels are normal. Corticosteroid therapy may help but bone marrow transplantation (BMT) may be the best hope.
Dental aspects
The face may be broad, with hypertelorism, snub nose and frontal bossing. There may be retarded tooth eruption. Radiography shows excessive bone density and thickness, especially obvious in the skull. Trigeminal or facial neuropathies may complicate the disease.
Fracture of the jaw or osteomyelitis may complicate dental extractions. Eruption of posterior teeth may be complicated by osteomyelitis. Once established, infection is difficult to eradicate, so that surgery should be minimally traumatic; mucoperiosteal flaps should preferably not be raised and antibiotic cover should be given. Management problems may include anaemia (Ch. 8), corticosteroid therapy (Ch. 6) or complications of BMT (Ch. 35).
Acquired Disorders
Rickets and osteomalacia
General aspects
Rickets is a disease of childhood characterized by inadequate skeletal mineralization. Osteomalacia has the same pathogenesis but affects adults, in whom there is failure of mineralization of replacement bone (osteoid) in the normal process of bone turnover. Both rickets and osteomalacia are largely related to a lack of vitamin D or calcium, due to the causes shown in Box 16.1. Rickets and osteomalacia are commonly results of a dietary deficiency in poorly nourished communities nowadays; South Asian immigrants living in northern Europe are particularly at risk. Contributory factors may be lack of sunshine exposure and eating chapattis made from wholemeal flour containing phytates, which impair calcium absorption.
Clinical features
In rickets, the bones are weak and readily deformed, fracturing easily but incompletely (greenstick or pseudofractures). Affected infants and young children are often also listless and irritable, with weak and hypotonic muscles, and they suffer bone pains. Excess formation of osteoid matrix causes a ‘rachitic rosary’ (swellings at costochondral junctions).
General management
Plasma alkaline phosphatase levels are usually raised, calcium levels low (in 50%), and phosphate levels normal or low. The underlying cause should be rectified where possible. Vitamin D and calcium should be given.
Dental aspects
Dental defects are seen only in unusually severe cases but eruption may be retarded. The jaws may show abnormal radiolucency. Vitamin D deficiency is not a contributory cause of dental caries.
In malabsorption syndromes, there may be secondary hyperparathyroidism or vitamin K deficiency, with endocrine or bleeding disorders, respectively, and oral manifestations of malabsorption.
In vitamin D-resistant rickets (familial hypophosphataemia), the skull sutures are wide and there may be frontal bossing. Dental complaints frequently bring attention to the disease, since the teeth have large pulp chambers and abnormal dentine calcification, and are liable to pulpitis and multiple dental abscesses. Since even minimal caries or attrition can lead to pulpitis, preventive care and fissure sealing or prophylactic occlusal coverage are needed.
Osteoporosis
General aspects
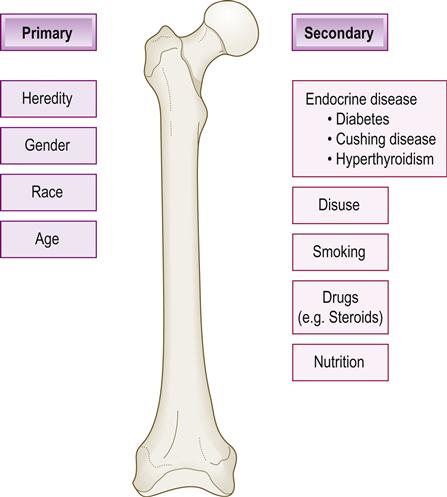
Osteoporosis is a very common condition of diminished bone mass and low bone density, which leads to fragile bones, usually in the elderly. Factors that lead to limited formation of bone early in life or loss of bone structure later in life lead to osteoporosis. The same factors that encourage bone formation in youth affect the maintenance of bone mass during the adult years, particularly calcium intake, reproductive hormone status, normal parathyroid gland function and physical activity (Fig. 16.4). The difference between how much healthy bone is formed during the first 28 or so years of life and the rate at which it is remodelled and removed later determines how much osteoporosis or osteopenia (see later) results. From age 30 years, about 1% of bone is lost per year, rising to about 5% after the menopause. During the first 20 years of life, formation of bone is the most important factor preventing osteoporosis, but after that point it is prevention of bone loss that becomes most important. Risk factors for osteoporosis are shown in Box 16.2.
Clinical features
Mildly traumatic or non-traumatic injuries in osteoporosis can cause fractures. The chief complications are fractures of the femoral neck, distal radius or humerus, or of the vertebral bodies (causing gradual collapse of the spine – hence the shrinking size of some older women). Low back pain is common.
General management
Diagnosis of osteoporosis is mainly from bone densitometry – a radiographic technique that gives accurate and precise measurements of the amount of bone (not the quality), termed ‘bone mineral density’ (BMD). Dual X-ray energy absorptiometry (DXA or DEXA) or sometimes dual photon absorptiometry (DPA) is used. BMD values that fall well below the average for the 25-year-old female (stated statistically as 2.5 standard deviations below the average) are diagnosed as ‘osteoporotic’. BMD values less than those in the normal 25-year-old female, but not 2.5 standard deviations below the average, are diagnosed as ‘osteopenic’. Initial screening for osteoporosis should be performed according to National Osteoporosis Foundation recommendations. The optimal interval for repeating DXA scans is uncertain, but because changes in bone density over short intervals are often smaller than the measurement error of most DXA scanners, frequent testing (e.g. at less than 2-year intervals) is unnecessary in most patients. Even in high-risk patients receiving drug therapy for osteoporosis, DXA changes do not always correlate with probability of fracture. Therefore, DXAs should only be repeated if the result will influence clinical management or if rapid changes in bone density are expected. Recent evidence also suggests that healthy women aged 67 and older with normal bone mass may not need additional DXA testing for up to 10 years, provided that osteoporosis risk factors do not change significantly. Poor BMD, raised serum alkaline phosphatase and urinary loss of calcium and hydroxyproline are risk markers but there are often no biochemical abnormalities. Bone biopsy is not practical or necessary.
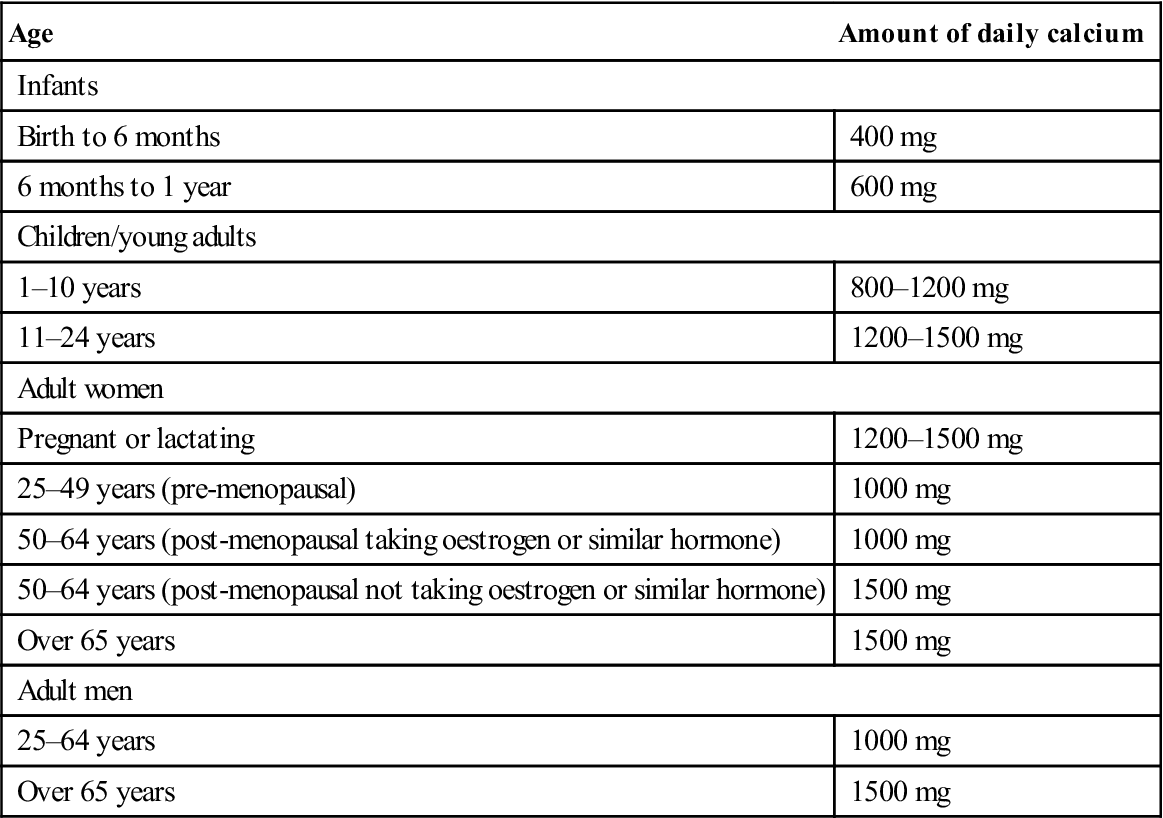
The best management is to avoid falls and injuries and to consider use of hip protectors. Exercise has a positive effect on BMD, particularly in adults who have been sedentary and begin to exercise. Weight-bearing exercises, such as walking, running, jogging and dancing, are recommended. Additional calcium and 1-alpha-hydroxyvitamin D may also be helpful. The main sources of calcium are dairy products and green vegetables; the main sources of vitamin D are sun exposure (15 minutes exposure daily from April to October in the northern hemisphere is sufficient), margarine, breakfast cereals, oily fish, eggs and meat. Table 16.2 shows the recommended calcium intake according to age, gender and hormone status.
Table 16.2
< ?comst?>
| Age | Amount of daily calcium |
| Infants | |
| Birth to 6 months | 400 mg |
| 6 months to 1 year | 600 mg |
| Children/young adults | |
| 1–10 years | 800–1200 mg |
| 11–24 years | 1200–1500 mg |
| Adult women | |
| Pregnant or lactating | 1200–1500 mg |
| 25–49 years (pre-menopausal) | 1000 mg |
| 50–64 years (post-menopausal taking oestrogen or similar hormone) | 1000 mg |
| 50–64 years (post-menopausal not taking oestrogen or similar hormone) | 1500 mg |
| Over 65 years | 1500 mg |
| Adult men | |
| 25–64 years | 1000 mg |
| Over 65 years | 1500 mg |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Hormone replacement therapy (HRT) may be effective in post- menopausal women and may also help relieve other symptoms, such as flushing (Ch. 25), but oestrogens can cause vaginal bleeding and increased risk of breast cancer. Raloxifene, given post-menopausally, may help to maintain BMD and assist the kidneys in producing vitamin D. Raloxifene also behaves like natural oestrogen in the liver, where it raises production of ‘good’ cholesterol (high-density lipoprotein; HDL) and lowers ‘bad’ cholesterol (low-density lipoprotein; LDL). Raloxifene appears to have no effect on the uterine endometrial lining growth, so there is no need for progestogen and there is no periodic bleeding. Raloxifene may also lower the breast cancer risk. The role of androgens (testosterone) in males is less clear, but the loss of testosterone promotes osteoporosis.
Anti-resorption drugs, which lower the rate at which osteoclasts reabsorb bone, include calcitonin, bisphosphonates and denosumab:
Strontium ranelate may inhibit osteoporosis but can cause serious drug rashes with eosinophilia and systemic symptoms (DRESS).
Dental aspects
There seems to be a correlation between osteoporosis and excessive alveolar bone loss in the elderly. Jaw osteoporosis is particularly a problem in women. Systemic treatment of osteoporosis may improve jaw bone density.
Patients with osteoporosis may have any of the problems of older people (Ch. 25). They may be at risk during general anaesthesia (GA) if there has been vertebral collapse and chest deformities.
Osteonecrosis of the jaw (ONJ; osteoclast modifier-related osteonecrosis of the jaws, OMRONJ)
Bisphosphonate-related or induced ONJ (BRONJ; BIONJ), defined as the presence of exposed bone in the mouth that fails to heal after appropriate intervention over a period of 6–8 weeks, has been reported in about 5% of patients with cancer who are receiving high-dose intravenous bisphosphonates. Use of oral bisphosphonates, such as alendronate and risedronate, results in an incidence of ONJ of about 1 case per 1500 patients to 2 cases per 100 000 patient years.
Bisphosphonates, especially if used intravenously, may induce osteonecrosis of the jaws. These drugs are used to treat not only osteopenia/osteoporosis but also hypercalcaemia of malignancy, metabolic bone disease (hyperparathyroidism, Paget disease) and osteogenesis imperfecta, Gaucher disease and Langerhans histio-cytosis. Bisphosphonates are pyrophosphate analogues that block the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase pathway (mevalonate path) and inhibit osteoclasts, thereby decreasing bone resorption. However, bisphosphonates remain in bone for years and have an extremely long-lasting effect. Bisphosphonate potency varies enormously, from the lowest potency (etidronate, clodronate and tiludronate) to the highest nitrogenous bisphosphonates (risedronate, ibandronate and zoledronate). Intravenous bisphosphonates such as pamidronate and zoledronate, used in cancer patients, are a particularly high risk for producing BIONJ (ranging from 1 in 10 to 1 in 100 patients). However, in osteoporosis, in which the doses used are an order of magnitude lower, the prevalence appears to be very much lower. Oral bisphosphonates such as alendronate used for more than 3 years carry a risk between 1 in 10 000 to 1 in 100 000, the lowest risk being with the lower-potency agents etidronate and tiludronate.
ONJ has also been seen in patients treated with other drugs used in cancer patients, such as denosumab, bevacizumab and sunitinib.
Some 67% of ONJ cases are preceded by tooth extraction, whereas another factor, such as a denture pressure sore or a torus, is identified in 7%; no predisposing factor is found in 26% of patients. The mean time to development of ONJ in cancer patients treated with zoledronate is 1.8 years but ranges from 6 to 60 months.
Preventive dentistry prior to initiation of high-dose antiresorptive therapy is important in patients with cancer.
A working party from US National Institutes of Health and Canadian Institutes of Health Research recommendations (Khosla et al., 2007) defined a confirmed case of BIONJ as:
an area of exposed bone in the maxillofacial region that did not heal within 8 weeks after identification by a health care worker, in a patient who was receiving or had been exposed to a bisphosphonate and had not had radiation therapy to the craniofacial region
and a suspected case as:
an area of exposed bone in the maxillofacial region that had been identified by a health care provider and had been present for<8 weeks in a patient who was receiving or had been exposed to a bisphosphonate and had not had radiation therapy to the craniofacial region.
Eighty per cent of cases present after surgery, as exposed necrotic bone, primarily mandibular alveolar bone (Fig. 16.5; Box 16.3). Lamina dura sclerosis or loss, and widening of the periodontal ligament space may well be early manifestations.
Prevention is by avoiding elective oral surgery; alternatively treatment should be carried out well before commencing bisphosphonates or after a drug holiday for 6 months. The patient must be counselled about risks and a risk assessment should be carried out. Risk factors include bisphosphonate type and dose (intravenous is worst), and dental co-morbidities (increase risk). Low bone turnover, as shown by low C-terminal cross-linking type 1 collagen telopeptides (CTX), increases risk. Recommendations for managing bisphosphonate patients are shown in Table 16.3.
Table 16.3
Recommendations for bisphosphonate patientsa
| Patients with non-malignant disease on bisphosphonates>3 y | Patients with malignancy, starting or already on bisphosphonates | |
| Surgery | Conventional orthograde endodontics are recommended rather than extraction where possible | Conventional orthograde endodontics are recommended rather than extraction where possible |
| Periodontics | Periodontal surgery is appropriate if it reduces or eliminates disease | No periodontal surgery |
| Endodontics | No apical surgery | No apical surgery |
| Implants | Currently not contraindicated if taking bisphosphonates but prudent to gain informed consent, which should be documented (risk assessment) | Not recommended |
If dental extractions become unavoidable in a patient taking bisphosphonates, the use of prophylactic antibiotics may be considered, but there are no controlled studies to support their use. The decision depends on the clinician’s level of concern relative to the individual patient and their specific situation, including concomitant risk factors (e.g. prolonged use of oral bisphosphonates, intravenous bisphosphonates, older age and concomitant use of oestrogen or glucocorticoids).
Therefore, whenever possible, dentists should avoid performing extractions and elective oral surgery in patients taking bisphosphonates. If surgery is essential, the dentist must counsel the patient about the risks of use of intravenous or oral bisphosphonates taken for more than 3 years. Teriparatide has recently been introduced for treatment of ONJ.
Osteoradionecrosis (ORN)
See Chapter 22.
Osteomyelitis
General aspects
Osteomyelitis literally means ‘inflammation of the bone marrow’, although sometimes the subperiosteal bone is mainly affected.
Haematogenous osteomyelitis is an infection caused by bacterial seeding from the blood, involves a single species of microorganism (typically a bacterium), occurs primarily in children, and is most common in the rapidly growing and highly vascular metaphysis of growing bones.
Direct or contiguous inoculation osteomyelitis is usually caused by trauma or surgery, and tends to involve multiple microorganisms.
Acute osteomyelitis of the jaws primarily affects the mandible, usually in adults, and is essentially an osteolytic destructive process, but an osteoblastic response is typical of sclerosing osteomyelitis. In children, proliferative periostitis may occur. Predisposing factors for osteomyelitis are sources from which bacteria can spread to bone, from:
■ local odontogenic infections (commonest cause)
■ other adjacent structures (e.g. otitis media, tonsillitis, suppurative sialadenitis)
■ haematogenous spread of organisms (rare in relation to the jaws).
When osteomyelitis of the mandible does occur, it may be a sign of an underlying debilitating disease, such as diabetes mellitus, an immune defect, an autoinflammatory condition or alcoholism. Alternatively, it may be related to reduced vascularity, such as after irradiation, or in rare bone conditions such as Paget disease or osteopetrosis.
Staphylococcus aureus is the commonest organism causing jaw osteomyelitis, but streptococci (both α- and β-haemolytic) and anaerobic organisms (e.g. Bacteroides and Peptostreptococcus species) are occasionally implicated. In acute osteomyelitis, the organisms excite acute inflammation in the medullary bone and the consequent oedema and exudation cause pus to be forced under pressure through the medullary bone, resulting in intrabone blood vessel (i.e. inferior dental artery) thrombosis, reducing the vascular supply to bone, which then necroses. Eventually, pus bursts through the cortical plate to drain via sinuses in the skin or mucosa. Where pus penetrates the cortex, it may spread subperiosteally, stripping the periosteum and thus further reducing the blood supply. Necrotic pieces of bone become sequestra, surrounded by pus, which either spontaneously discharge or remain and perpetuate infection. The periosteum lays down new bone to form an involucrum encasing the infected and sequestrated bone. The involucrum may prevent sequestra from being shed. Finally, if little new bone is formed, a pathological fracture may occur.
Clinical features
Jaw osteomyelitis presents with deep-seated, boring pain and swelling. Teeth in the affected area become mobile and tender to percussion, with pus oozing from the gingival crevices. The pain abates once pus penetrates the cortical plate, and discharges intraorally or extraorally, often through several sinuses. Labial anaesthesia is a characteristic feature because of pressure on the inferior alveolar nerve. The patient is often febrile and toxic with enlarged regional lymph nodes.
General management
Diagnosis is clinical, supported by imaging that, in established cases, shows marked bony destruction and sequestration. However, since radio-graphic changes are seen only after there has been significant decalcification of bone, early cases may not be detected; in these, isotope bone scanning using technetium diphosphonate may show increased uptake. Blood tests show a leukocytosis with neutrophilia and a raised erythrocyte sedimentation rate (ESR).
Treatment is with antibiotics and usually drainage. Penicillin was the drug of choice, but since many staphylococci are now penicillin-resistant, flucloxacillin or fusidic acid may be used. Lincomycin and clindamycin also give high bone levels but, because of a high incidence of pseudomembranous colitis, are now regarded as second-line drugs. Metronidazole gives good bone levels and is indicated for anaerobic infections. Drainage of pus follows the guidelines used for other infections. When dead bone is exposed in the mouth, it can often be left to sequestrate without surgical interference, but sequestrectomy should be undertaken if the separated dead bone fails to discharge; decortication of the mandible may be needed in order to allow this, along with drainage. Hyperbaric oxygen is an effective adjunct for recalcitrant osteomyelitis, especially where anaerobic organisms are involved.
Acute maxillary osteomyelitis is rare, and usually seen in infants, presumably because the lack of development of the antrum at this age means the maxilla is a dense bone.
Chronic osteomyelitis presents with intermittent pain and swelling, relieved by the discharge of pus through long-standing sinuses. Bone destruction is localized and often a single sequestrum may be the source of chronic infection. Removal of the sequestrum and curettage of the associated granulation tissue usually produces complete resolution.
Focal sclerosing osteomyelitis is usually asymptomatic and is revealed as an incidental radiographic finding. Most common in young adults, there is a dense, radio-opaque area of sclerotic bone caused by formation of endosteal bone – usually related to the apical area of a mandibular molar. This appears to be a response to low-grade infection in a highly immune host. Following tooth extraction, the infection usually resolves (but the sclerotic bone often remains).
Diffuse sclerosing osteomyelitis is a sclerotic endosteal reaction, which, like focal sclerosing osteomyelitis, appears to be a response to low-grade infection. However, the area of bone involved is widespread and sometimes involves most of the mandible or occasionally the maxilla. Sometimes the infection arises in an abnormally osteosclerotic mandible, such as in Paget disease, osteopetrosis or fibrous dysplasia. Intermittent swelling, pain and discharge of pus may go on for years. Management is difficult because of the extensive nature of the disease process. Long-term antibiotics, curettage and limited sequestrectomy all have their place.
Proliferative periostitis (Garré osteomyelitis) is more common in children than adults. The cellular osteogenic periosteum of the child responds to low-grade infection, such as apical infection of a lower first molar tooth, by proliferation and deposition of subperiosteal new bone. The subperiosteal bone may be deposited in layers, producing an onion-skin appearance radiologically, which can simulate Ewing sarcoma. The endosteal bone, however, may appear to be completely normal, but in severe cases also appears moth-eaten radiologically. Removal of the infective source is usually followed by complete resolution, although subsequent bone remodelling can take a considerable time.
Tumoral calcinosis
This rare disease is characterized by ectopic, soft tissue calcifications but normocalcaemia. Early-onset periodontitis may be a feature.
Fibrous dysplasia
General aspects
Fibrous dysplasia consists of replacement of an area of bone by fibrous tissue, causing a localized swelling that may affect a single bone (monostotic) or, less commonly, several bones (polyostotic).
The aetiology of fibrous dysplasia is unknown. The process typically starts in childhood and, as the skeleton matures, the lesions ossify progressively; ultimately, when skeletal growth ceases, they usually become stabilized.
Cherubism shares some features in common with fibrous dysplasia but may cause bilateral lesions of the maxilla causing the eyes to ‘up-turn towards heaven’.
Clinical features
Monostotic fibrous dysplasia is the most common type; it is more frequent in females and often involves the jaws, particularly the maxilla. Polyostotic fibrous dysplasia may affect either sex and may involve over 50% of the skeleton, but is uncommon. The lesions may be unilateral and 50% have abnormal skin hyperpigmentation of a café-au-lait colour, especially over the dorsum of the trunk and limbs, usually on the same side as the bone lesions (Jaffe syndrome). Albright syndrome is polyostotic fibrous dysplasia with skin hyperpigmentation and precocious puberty in females; there are episodic rises in serum oestrogen and falls in gonadotropin levels.
General management
Radiography typically shows a ground-glass appearance with no defined margins. Serum calcium and phosphate levels are normal, often with raised serum alkaline phosphatase level and raised urinary hydroxyproline.
Fibrous dysplasia is typically self-limiting and usually ceases to progress after adolescence. Patients are sometimes managed with calcitonin. Bisphosphonate treatment may have implications (see Osteonecrosis of the jaw). Testolactone is effective for the precocious puberty of Albright syndrome. Surgery can be used to correct any residual cosmetic defect but the bone tends to bleed a great deal. Radiotherapy should be avoided because of the risk of inducing osteosarcomas.
Dental aspects
The facial bones are frequently involved in monostotic fibrous dysplasia and in 25% of cases of polyostotic disease. Monostotic fibrous dysplasia is not a medical problem but it is possible for the polyostotic type of the disease to be unrecognized when the facial lesions are the most obvious feature. Hyperthyroidism or diabetes mellitus may occasionally be associated with polyostotic fibrous dysplasia and therefore may, rarely, complicate treatment. Mucosal pigmentation may be seen but is rare in Albright syndrome. In the differential diagnosis, it may be confused with that of Addison disease, while the skin pigmentation may simulate that of neurofibromatosis.
Paget Disease of Bone (Osteitis Deformans)
General aspects
Paget disease is characterized by progressive deformity and enlargement of bones related to overactivity of osteoclasts and osteoblasts. There appears to be considerable geographical variation in the prevalence, disease being common in northern Europeans, and also in older age. Paget disease is rare in Africans, people from the Indian subcontinent, and other Asians.
The essential features are total disorganization of the normal orderly replacement of bone, and an anarchic alternation of bone resorption and apposition. In the earlier stages, resorption predominates (osteolytic phase) but later bone apposition takes over and, as disease activity declines, the affected bones become enlarged and dense (sclerotic stage).
There is a male predominance. The aetiology of Paget disease is unknown and a slow virus has been implicated. Inherited forms of Paget disease of bone, caused by mutations in genes such as SQSTM1 that affect osteoclast differentiation and function, are recognized.
Clinical features
Symptoms are absent in the early stages, which can only be detected by imaging. Bone pain is the most common feature in older patients; it is usually felt around the hips or knees but is non-specific. One or many bones may be affected. In the polyostotic type, the axial skeleton is mainly involved but hands and feet are usually spared (Fig. 16.6). Other possible complications of Paget disease are deformities, pathological fractures, and constriction of skull foraminae – sometimes compressing cranial nerves and leading to effects such as deafness or impairment of vision. Rarely, there is brainstem or spinal compression.
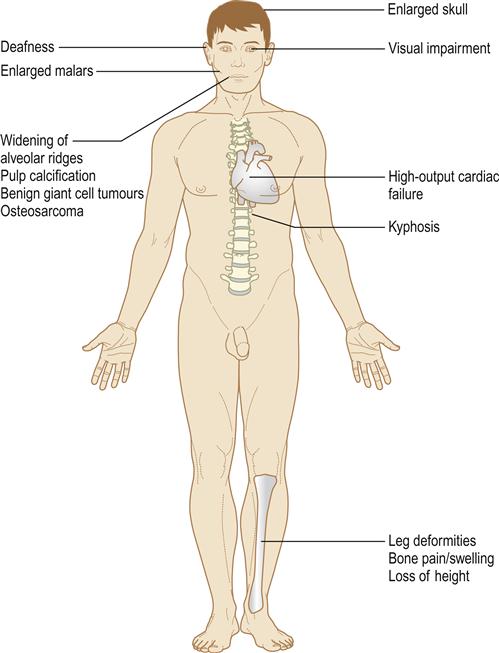
If Paget disease is widespread, hypervascularity of the bones can produce, in effect, an arteriovenous fistula and high-output cardiac failure. Development of osteosarcoma is a recognized but rare complication.
General management
The differential diagnosis includes hyperostosis frontalis interna (a benign condition characterized by frontal bone sclerosis), fibrous dysplasia, pustulotic arthrosteitis (sclerotic lesions of clavicle and ribs) and osteosclerotic metastases. Diagnosis is from radiographs of affected bones, which show predominantly radiolucencies due to osteolysis early on but mixed areas of lysis with sclerosis later. Affected bones become thicker. Radionuclide bone scanning showing increased uptake of technetium diphosphonate helps define disease extent. The serum total alkaline phosphatase level is grossly raised when many bones are affected, but usually there is little or no change in calcium or phosphate levels. More specific is a rise in bone-specific alkaline phosphatase, procollagen type I N-terminal propeptide and osteocalcin. There is also raised urinary hydroxyproline and pyridinoline excretion. Biopsy is rarely indicated.
Currently, the most effective treatment of Paget disease is with bisphosphonates. Vitamin D deficiency is a common accompaniment.
Dental aspects
When the skull is affected, a typical early feature is a large irregular area of relative radiolucency (osteoporosis circumscripta; Fig. 16.7). Later, there is radio-opacity, with loss of the normal landmarks and an irregular cotton-wool appearance. Basilar invagination may be seen. The maxilla is involved occasionally and the mandible rarely. Typically, maxillary enlargement causes symmetrical malar bulging (leontiasis ossea; Fig. 16.8). The intraoral features are gross symmetrical widening of the alveolar ridges, loss of lamina dura and hypercementosis, often forming enormous craggy masses that may become fused to the surrounding bone. Pulpal calcification may be seen. Serious complications follow efforts to extract severely hypercementosed teeth. Attempts using forceps may fail to move the tooth or cause fracture of the alveolar bone. Alternatively, the tooth may be mobilized but retained, as if in a ball-and-socket joint. If hypercementosis is severe, a surgical approach with adequate exposure should be used for extractions. In the early stages of the disease, the highly vascular bone may bleed freely; later, the poor blood supply to the bone makes it susceptible to chronic suppurative osteomyelitis. Bisphosphonate treatment may have implications (see Osteonecrosis of the jaw). Prophylactic antibiotics, such as penicillin (immediately before and for 3 or 4 days after the operation), may help prevent postoperative infection. Dentures have to be replaced as the alveolar ridge enlarges. Benign giant cell tumours may be seen.
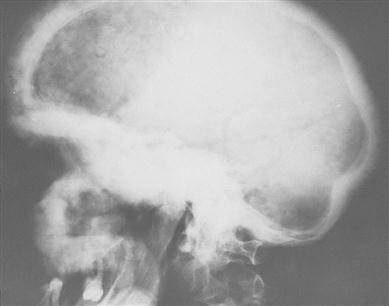
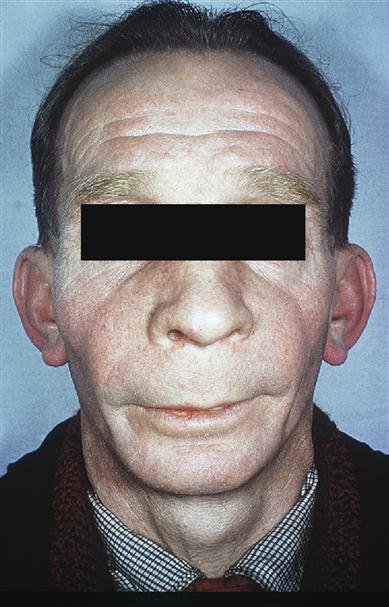
Patients with Paget disease involving facial or maxillo-mandibular parts of the skeleton have a higher prevalence of heart disease and of deteriorating hearing, sight and smell. Osteosarcoma is rare in the jaws. />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


