CHAPTER 16 Local Anesthetics
CHEMISTRY AND CLASSIFICATION
Structure-Activity Relationships
The typical local anesthetic molecule can be divided into three parts: (1) an aromatic group, (2) an intermediate chain, and (3) a secondary or tertiary amino terminus (Figure 16-1). All three components are important determinants of a drug’s local anesthetic activity. The aromatic residue confers lipophilic properties on the molecule, whereas the amino group furnishes water solubility. The intermediate portion is significant in two respects. First, it provides the necessary spatial separation between the lipophilic and hydrophilic ends of the local anesthetic. Second, the chemical link between the central hydrocarbon chain and the aromatic moiety serves as a suitable basis for classification of most local anesthetics into two groups, the esters (–COO–) and the amides (–NHCO–). This distinction is useful because there are marked differences in allergenicity and metabolism between the two drug categories.
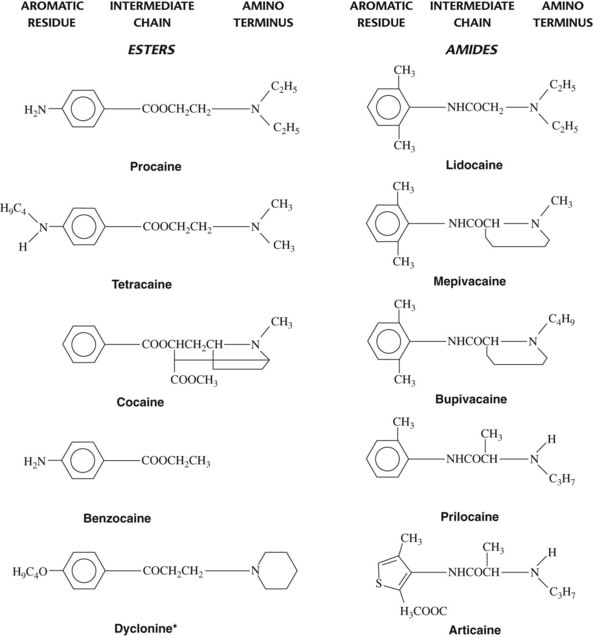
Minor modifications of any portion of the local anesthetic molecule can significantly influence drug action. The addition of a chlorine atom to the ortho position on the benzene ring of procaine yields chloroprocaine, a more lipophilic local anesthetic four times as potent as the parent compound yet half as toxic when injected subcutaneously. Table 16-1 lists several important physicochemical properties of local anesthetics and how they correlate with clinical activity.
Influence of pH
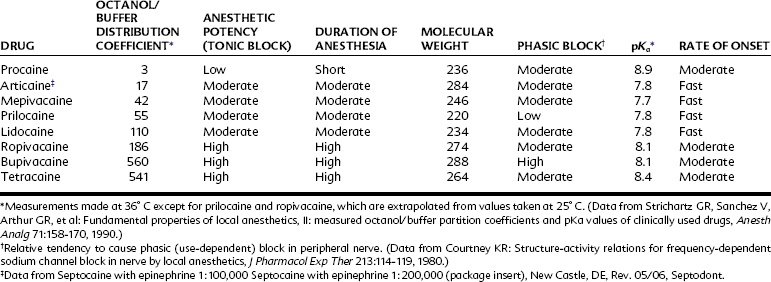
By virtue of the substituted amino group, most local anesthetics are weak bases with a negative logarithm of the acid ionization constant (pKa) ranging from 7.5 to 9.0. A local anesthetic intended for injection is usually prepared in salt form by the addition of hydrochloric acid. Not only is water solubility improved, but also stability in aqueous media is increased. When injected, the acidic local anesthetic solution is quickly neutralized by tissue fluid buffers, and a fraction of the cationic form is converted to the nonionized base. As determined by the Henderson-Hasselbalch equation (Figure 16-2), the percentage of drug converted depends primarily on the local anesthetic pKa and the tissue pH. Because only the base form can diffuse rapidly into the nerve, drugs with a high pKa tend to be slower in onset than similar agents with more favorable dissociation constants. Tissue acidity may also impede the development of local anesthesia. Products of inflammation can lower the pH of the affected tissue and limit formation of the free base. Ionic entrapment of the local anesthetic in the extracellular space delays the onset of local anesthesia and may render effective nerve blockade impossible.
An alternative approach to modifying drug distribution is through the addition of carbon dioxide. Carbonation of a local anesthetic solution can increase the rate of onset and sometimes the depth of anesthesia. It has been suggested that the hydrocarbonate salt of the local anesthetic penetrates membranes more rapidly than the conventional formulation and that the injected carbon dioxide diffusing into the nerve trunk lowers the internal pH and concentrates local anesthetic molecules by ion trapping.55 There is also evidence that carbon dioxide may potentiate local anesthetic activity by a direct effect on the nerve membrane.15,19 Although promising, carbonated local anesthetic solutions are unavailable in the United States, and a study of carbonated lidocaine used for mandibular anesthesia failed to reveal any significant benefit compared with lidocaine hydrochloride.22
MECHANISM OF ACTION
Effects on Ionic Permeability
The quiescent nerve membrane is impermeable to Na+. Excitation of the neuron by an appropriate stimulus temporarily increases Na+ conductance and causes the nerve cell to become less electronegative regarding the outside. If the transmembrane potential is sufficiently depressed, a critical threshold is reached at which the depolarization becomes self-generating. Local electrotonic currents induce a rapid influx of Na+ through activated Na+-selective channels traversing the nerve membrane. The inward Na+ current creates an action potential of approximately +40 mV, which is propagated down the nerve. The action potential is quite transient at any given segment of membrane; loss of Na+ permeability (inactivation of the Na+ channels) and an outward flow of K+ (in nonmyelinated axons) quickly repolarize the membrane. These events are reviewed in Figure 16-3.
Local anesthetics interfere with nerve transmission by blocking the influence of stimulation on Na+ conductance. A developing local anesthetic block is characterized by a progressive reduction in the rate and degree of depolarization and a slowing of conduction. When the depolarization is retarded sufficiently such that repolarization processes develop before the threshold potential can be reached, nerve conduction fails.1
Site of Action
Several sites exist within the nerve membrane where drugs could potentially interfere with Na+ permeability. It was argued that local anesthetics could interact with membrane lipids to impair Na+ channel function, just as had long been proposed for general anesthetics (see Chapter 17).86 In recent years, evidence has accumulated that conventional local anesthetics interact directly with Na+ channels to inhibit nerve conduction.18,88 Each Na+ channel is composed of several subunits. The α subunit is the largest component (260 kDa) and forms the actual channel,20 whereas the smaller β subunits help to stabilize the channel complex within the membrane.56 As depicted in Figure 16-4, the α subunit consists of four homologous domains (I to IV), each of which is composed of six structurally similar helical segments (S1 to S6) that traverse the plasma membrane. Collectively, the S4 segments of each domain constitute the voltage sensor of the “m” or “activation” gate, which opens in response to a depolarizing stimulus. Each S4 segment contains positively charged amino acid residues, specifically arginine and lysine, at every third position of the α helix. In the “helical screw model” of activation,21 depolarization causes the outward conformational rotation of the S4 segments, which can be detected experimentally as the small gating currents that precede the action potential. Local anesthetic blockade of the Na+ channel is characterized by a reduction in the peptide movements responsible for these gating currents.63 Lidocaine tends to trap the S4 segment of domain III in the external, depolarized configuration and to retard movements of the S4 segment of domain IV.77 As a consequence, the Na+ channel remains in an inactivated configuration that precludes normal opening.
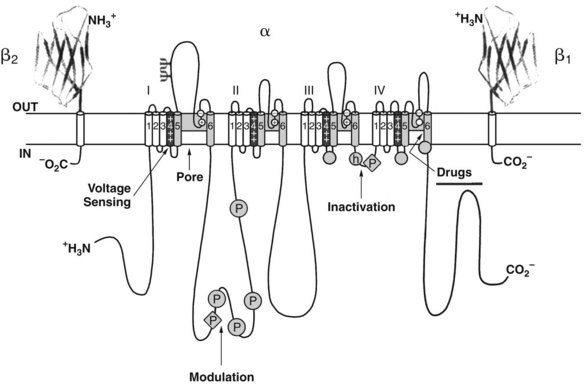
As the active site for local anesthetics resides within the Na+ channel, access becomes an important issue. In this regard, studies with permanently charged local anesthetics have proved enlightening.44 Conversion of the amino terminus of certain local anesthetics (e.g., lidocaine) to the quaternary form (e.g., QX-314) yields permanently charged cations largely incapable of crossing the nerve membrane. Although ineffective when applied externally to the axolemma, these experimental compounds show full blocking activity on internal administration. They gain access to the receptor by traveling up an aqueous route within the Na+ channel, which must be fully open or at least partially activated to permit their entry from the cytoplasm. Lipophilic molecules, such as benzocaine or the uncharged form of lidocaine, can reach the channel and receptor site by traversing a hydrophobic route, which may include the membrane lipid and hydrophobic portions of the Na+ channel.
Specific mutations of the S6 segment of domain IV of the Na+ channel greatly alter local anesthetic blockade.68 Replacement of the phenylalanine amino acid midway down the S6 helix with an alanine residue reduces by 99% the apparent binding affinity of the local anesthetic etidocaine to open and inactivated channels. A similar, although smaller, effect occurs when the tyrosine located 11 Å and two turns inward on the same side of the S6 helix is replaced with alanine. Because these aromatic amino acids can interact with local anesthetics through hydrophobic and van der Waals interactions, and their spatial separation conforms to the length of the typical local anesthetic molecule (10 Å to 15 Å), they are believed to be part of and to identify the local anesthetic receptor site. Mutation studies have also shown specific amino acid residues on the S6 segments of domains I and III that appear to form part of the receptor site (Figure 16-5).94
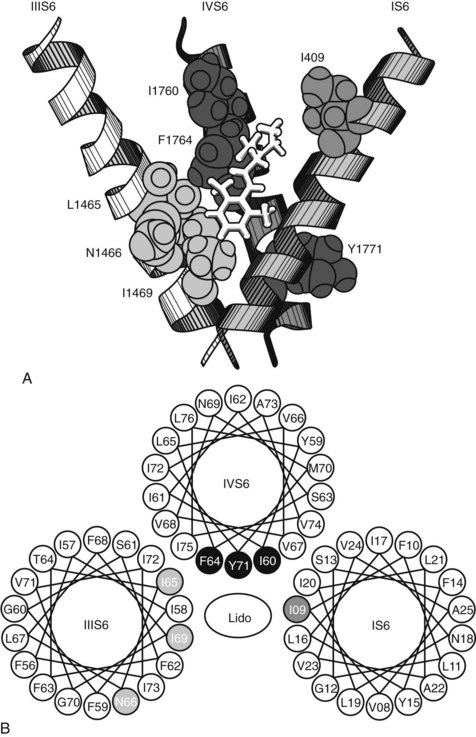
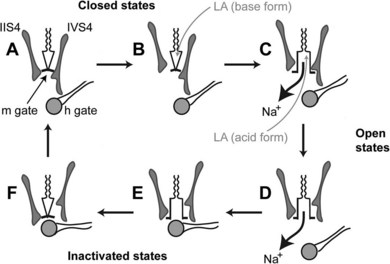
As previously mentioned, local anesthetics block nerve conduction by impeding the gating mechanisms that underlie cycling of the Na+ channel. Other actions that could contribute to nerve blockade include a physical occlusion of the channel, an allosterically mediated change in channel conformation, and (at least with local anesthetic cations) a distortion of the local electrical field.57 Some of these actions may be complementary, as binding of a cationic local anesthetic molecule to the putative receptor site places its positive charge adjacent to the most constricted portion of the channel and stabilizes the S4 domain III segment (and to a lesser degree the S4 domain IV segment) in the extruded position.62 Figure 16-6 depicts the Na+ channel as it cycles through its primary configurations in response to a depolarizing stimulus and postulated interactions with neutral and charged local anesthetic species.5
Use-Dependent Block
Numerous studies have proved that high-frequency stimulation increases the magnitude of channel blockade by local anesthetics. Within certain limits, the degree of axonal block is strongly and continuously dependent on the stimulus rate regardless of equilibration time. A concentration of lidocaine that reduces a compound action potential by 40% at a stimulation rate of 1 Hz causes a depression of 80% after 15 seconds at 40 Hz.14 These results, as embodied in the modulated receptor hypothesis of local anesthesia, suggest that stimulation of the nerve membrane not only exposes the site of action to local anesthetic cations, but also increases temporarily the affinity of the channel receptor for them.
As originally proposed by Hille,44 the modulated receptor hypothesis holds that both the charged and neutral forms of local anesthetics bind preferentially to open and inactivated Na+ channels. The binding reciprocally tends to stabilize the channels in the inactivated state. If stimulations are sufficiently infrequent, time is available after each depolarization for the slower-than-normal transition from inactivated to resting channels to take place. This conversion reduces local anesthetic binding and permits a net diffusion of neutral anesthetic molecules out of the channels. The remaining anesthetic bound to closed channels provides a basal or tonic block. Conversely, repeated stimuli do not allow for full recovery between depolarizations; anesthetic binding remains enhanced, Na+ channels in the inactive configuration accumulate, and use-dependent block ensues. Subsequent refinements to the modulated receptor hypothesis include discoveries that the increased affinity state of the receptor caused by depolarization of the membrane is not synonymous with the classically defined open or inactivated forms of the channel but may include closed but partially activated channels and several “slow” inactivated configurations promoted by local anesthetic binding.
Marked differences in use dependency have been recorded for various local anesthetics.24 Benzocaine and related nonionized compounds show little phasic block and then only at very high stimulus rates. Conventional local anesthetics exhibit an approximate 10-fold range in frequency dependence, with phasic block becoming clinically significant at 2.5 Hz for lidocaine and at 0.5 Hz for bupivacaine. Permanently charged local anesthetic derivatives develop use-dependent blocks with stimulus rates of 2.4 per minute (0.04 Hz). Basic knowledge gained by the study of use dependency is increasingly being applied to clinical questions involving local anesthetic efficacy and toxicity and to related classes of drugs, such as various antiarrhythmic and anticonvulsant agents that also exhibit phasic block. Ultimately, new drugs and modes of therapy are expected to arise from the pharmaceutical exploitation of this phenomenon.
Differential Nerve Block
Critical length
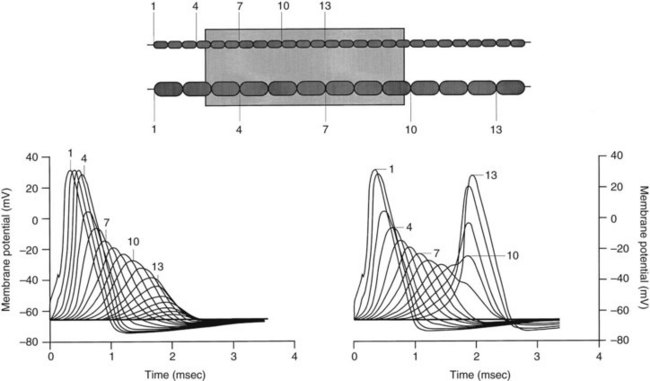
The clinical observations already described (and best seen after spinal or epidural anesthesia) should not be construed as proof that large myelinated axons are inherently more resistant to local anesthetics than smaller fibers. A careful study of individual axons by Franz and Perry35 revealed that the minimum blocking concentration of procaine is not directly related to fiber diameter. A differential block, in which small C and A fibers were affected but larger A fibers were not, could be obtained but only when the length of compound nerve exposed to procaine was restricted in length. On the basis of these findings, the authors concluded that differential sensitivities of fibers of unequal diameter result from variations in the “critical length” that must be exposed to a local anesthetic for conduction to fail.
When the concentration of local anesthetic is insufficient to block three adjacent nodes completely, anesthesia may still occur if a larger train of nodes is partially blocked.34,69 As long as more than 70% of the Na+ channels in a node are inhibited, the resulting action potential at that node is reduced in size. Progressive declines in the action potentials of partially blocked nodes along the axon ultimately result in failure of conduction if a sufficient length of nerve is exposed to the drug. As shown in Figure 16-7, smaller neurons are again more readily blocked because of the shorter length required for exposure of the requisite number of nodes.69
Use-dependent block
In addition to anatomic and physiologic variables, the pattern of impulse traffic normally carried in situ by the different nerve fibers may contribute greatly to a differential nerve block.73 Noxious stimuli and sympathetic nervous system transmissions are encoded in rapid bursts of impulses, whereas motor function usually involves low-frequency discharges. Local anesthetics whose use-dependent characteristics fall within this frequency range tend to block pain sensations and autonomic responses preferentially.
Local anesthetic selectivity
Local anesthetics vary in their relative inherent ability to block sensory versus motor fibers. A good example of this form of differential block involves bupivacaine and etidocaine. Both of these drugs are highly lipid-soluble agents capable of producing prolonged nerve blockade. Bupivacaine can elicit sensory anesthesia at one third the concentration required for motor blockade, whereas etidocaine shows no selectivity of effect.79 Because maintenance of uterine muscle contractility is important in childbirth, bupivacaine is the preferred agent for epidural anesthesia during labor and delivery.
The mechanism behind such differential effects of local anesthetics has not been elucidated. One possibility relates to the drugs’ relative tendency to block different K+ channel subtypes. A local anesthetic (presumably etidocaine) with a strong ability to block voltage-gated K+ channels important in reversing neuronal depolarization might be expected to work against itself in nonmyelinated axons subserving the perception of pain. In such nerves, inhibition of K+ efflux would give Na+ efflux a better chance of reaching threshold and propagating the action potential. A selective blockade of K+ influx through K+ channels that specifically control the resting membrane potential of small, nociceptive axons would result in partial membrane depolarization and a potentiation of Na+ channel inactivation and local anesthetic blockade.49
Inflammation
The failure to obtain satisfactory clinical pain relief in inflamed tissues is a well-known and undesirable form of differential nerve block. Clinically, this phenomenon is encountered in a patient who exhibits profound local anesthetic effect except in the specific area requiring treatment. If inflammation lowers the pH at the injection site, diffusion of the drug into the axolemma would be impaired, as described previously. There is some evidence, however, that the buffering capacity of inflamed tissues is not always reduced67 and that other reasons for local anesthetic failure must exist in these conditions.
Increased blood flow and decreased catecholamine effectiveness in inflamed tissues may speed removal of the local anesthetic from the injection site. Alteration of Na+ channel number, function, or type may offset the ability of local anesthetics to block nerve conduction. Analogous changes in the expression or activity of other ion channels involved in nociception may have a similar effect.52 Neuromediators and other products released or synthesized during inflammation may increase responsiveness of nociceptors and/or enhance nerve conduction in response to painful stimuli. These include histamine, prostaglandin E1, kinins, adenine nucleotides, and substance P.
PHARMACOLOGIC EFFECTS
Central Nervous System
Sensitive psychomotor tests and subjective reports of mild drowsiness indicate that systemic effects caused by local anesthetics can occur with plasma concentrations that are achieved in dental patients.6 Analgesic and anticonvulsant effects also occur in subtoxic concentrations. Initial signs and symptoms of a toxic effect are often excitatory in nature and consist of a feeling of lightheadedness and dizziness, followed by visual and auditory disturbances, apprehension, disorientation, and localized involuntary muscular activity. Depressant responses, such as slurred speech, drowsiness, and unconsciousness, may also occur and are especially prominent with certain drugs (e.g., lidocaine). As higher blood concentrations of drug are attained, muscular fasciculations and tremors intensify and develop into generalized tonic-clonic convulsions. On termination, seizure activity is often succeeded by a state of CNS depression identical to general anesthesia. With excessively large doses, respiratory impairment becomes manifest; if untreated, death by asphyxiation may ensue.
The CNS excitation sometimes observed after local anesthesia is intriguing because the sole action ascribed to these agents is one of depression. Studies involving the topical application of local anesthetics to exposed cortical or spinal cord neurons document that the only direct effect of procaine and related drugs is to inhibit electrical activity.26 The apparent stimulation observed clinically may be explained on the basis that inhibitory cortical neurons or synapses are highly susceptible to transmission block. Initial disruption of these pathways results in a disinhibition of excitatory neurons, manifested clinically as stimulation. Electroencephalographic studies indicate that local anesthetic seizures begin in the amygdala.37,76 Disinhibition of this part of the limbic system allows high-voltage discharges to occur, which spread throughout the brain. A more recent finding that local anesthetics can block a family of K+ channels (whose inhibition increases neuronal excitability) raises the possibility that CNS stimulation and cardiac arrhythmias may arise in part from direct neuronal excitation.49
Cardiovascular System
Myocardium
Presumably because of their ability to block Ca++ channels and evoked Ca++ release from the sarcoplasmic reticulum and to reduce myofibrillar responsiveness to available Ca++, local anesthetics depress myocardial contractility in a dose-dependent manner.59 With conventional doses of lidocaine, this effect is minor and sympathetic reflexes and direct vascular effects produce a compensatory increase in peripheral resistance, which prevents a decrease in blood pressure. Through a centrally mediated disinhibition of sympathetic nervous activity, heart rate and arterial blood pressure may become elevated coincident with CNS excitation. Conversely, mepivacaine has been reported in moderate doses to decrease peripheral vascular resistance and increase cardiac output,48 which suggests that local anesthetics may exert dissimilar patterns of direct and indirect effects on the heart at subtoxic blood concentrations.
Reports in humans suggest and experiments in several other species confirm that bupivacaine and certain other highly lipophilic local anesthetics are cardiotoxic compared with less lipophilic congeners. Serious ventricular arrhythmias and cardiovascular collapse are more likely to occur, and resuscitation is more problematic. One explanation for these observations involves use-dependent blockade.24 As indicated in Table 16-2, bupivacaine has a high molecular weight for a local anesthetic. That, coupled with its lipophilic tendency and perhaps its high pKa, enables the drug to exert a strong phasic block at normal heart rates. Inhibition of K+ and Ca++ channels/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


