CHAPTER 14 Anticonvulsants*
CLASSIFICATION OF EPILEPTIC DISORDERS
The classification proposed in 1989 by the Commission on Classification and Terminology of the International League Against Epilepsy (ILAE) is complex because of the variable characteristics of many epileptic syndromes. A simplified approach more suited to this discussion limits consideration to the seizures themselves (Table 14-1). Seizure patterns are broadly divided into two major groups: (1) partial seizures, in which convulsions begin in a localized region of the brain, involve restricted areas of the body, are initially unilateral, and yield EEG recordings of rhythmic activity that is restricted at least initially to one hemisphere, and (2) generalized seizures, with convulsions often involving the entire body and EEG recordings having characteristic bilateral patterns. A modified ILAE classification scheme is under development to classify epilepsy based on Axis 1, descriptive terminology for ictal events; Axis 2, seizure description; Axis 3, syndromes and diseases; and Axis 4, life impairment.10
| CLASSIFICATION | CLINICAL ASPECTS |
|---|---|
| I. Partial (focal, local) seizures | Involves one side of brain at onset |
| A. Simple partial seizures (e.g., Jacksonian) | Consciousness not impaired; specific or localized motor, sensory |
| B. Complex partial seizures (e.g., psychomotor, temporal lobe) | Consciousness impaired, automatisms, autonomic or psychological signs or symptoms; patients may report aura beforehand |
| C. Partial seizures evolving to generalized seizures | See generalized seizures; patients may report aura beforehand |
| II. Generalized seizures | Involve both sides of brain at onset |
| A. Tonic-clonic seizures (grand mal) | Consciousness is lost; bilateral sharp tonic contraction of muscles, generalized from onset, followed by clonic contractions; patient may report aura before seizure |
| B. Absence seizures (e.g., petit mal) | Consciousness impaired, postural muscles not impaired, EEG spike and slow wave complexes at approximately 3 Hz |
| C. Myoclonic seizures | Sudden, brief contractions of individual muscles or groups producing shocklike spasms in muscles of face, trunk, and extremities |
| D. Clonic seizures | Repetitive clonic jerking (alternating contractions of opposing muscles) |
| E. Tonic seizures | Violent muscular contraction (simultaneous contraction of flexors and extensors) with limbs in strained position |
| F. Atonic seizures (astatic) | Sudden loss of muscle tone, consciousness sometimes lost, patients sustain fall injuries |
| III. Unclassified seizures | Cannot be classified because of insufficient data or atypical pattern of seizure |
EEG, Electroencephalogram.
Adapted from Commission on Classification and Terminology of the International League Against Epilepsy, Epilepsia 22:489–501, 1981.
Generalized Seizures
Generalized seizures occurring in the form of repeated or continuous attacks are referred to as status epilepticus. Tonic-clonic status epilepticus is rare but life-threatening. Status epilepticus may develop in patients with convulsive disorders, with acute disease affecting the brain (meningitis, encephalitis, toxemia of pregnancy, uremia, acute electrolyte imbalances), after abrupt withdrawal of depressant or anticonvulsant medication (barbiturates, benzodiazepines, opioids), or rarely after local anesthetic administration. Status epilepticus can occur in the absence of a prior history of seizures. The drugs most widely used to treat status epilepticus are intravenous benzodiazepines (lorazepam, diazepam, and midazolam), phenytoin, fosphenytoin, phenobarbital, and valproic acid.17,35,48 In refractory status epilepticus, the patient may have to undergo general anesthesia (e.g., midazolam, propofol, thiopental, and pentobarbital). An anesthetic dose of pentobarbital or propofol is effective and has a more rapid onset than phenobarbital. Because large doses of these drugs are usually required, there is the danger of respiratory depression and respiratory arrest, especially with barbiturates or propofol. Grand mal status epilepticus is best treated in a hospital setting.
PATHOPHYSIOLOGY
The pathophysiologic characteristics of epilepsies are not well understood. Idiopathic epilepsy has a primary genetic basis, with some influence of environmental factors.26 The various types of epilepsies share many features but also differ in many respects. The fact that many anticonvulsant drugs are selective for specific seizure types12 suggests that the origin and progression of all seizures are not identical. Several hypotheses have been proposed to explain why seizures occur. These hypotheses focus on defects in (1) ionic conductance of the neuronal membrane, including Na+, Ca++, K+, Cl−, and H+; (2) inhibitory neuronal circuits, especially those involving the inhibitory neurotransmitter γ-aminobutyric acid (GABA); (3) excitatory mechanisms, especially those involving the excitatory neurotransmitter glutamate; (4) altered synaptic function; (5) depressed energy metabolism; and (6) other processes supporting presynaptic or postsynaptic function, such as other neurotransmitters with modulatory roles, peptides, hormones, growth factors, second messengers, nuclear changes, glial function, and gap junctional function.
Different brain structures may participate as seizure sources. The cortex is often involved. In complex partial epilepsy, unusual activity in the temporal lobe and limbic structures is found. A more recent gene chip study identified abnormal release of glutamate from astrocytes as a significant change in temporal lobe epileptic foci.28 For absence seizures, changes in the thalamus, basal ganglia, and substantia nigra pars reticulata may be involved.50 Audiogenic seizures seem to involve the mesencephalon and basal ganglia.
Diagnostic imaging is being used to help localize the sites of abnormal brain function in epilepsy. Positron labeled 2-[18F]fluoro-2-deoxy-d-glucose has been approved as an aid for diagnosis of epilepsy by PET. Generally, epileptic zones show hypometabolism in the intraictal state. Another PET imaging technique involves the use of the benzodiazepine antagonist flumazenil, which visualizes generally decreased binding in epileptic tissues.40 Additional tracers and imaging techniques are being developed.
ANTICONVULSANT THERAPY
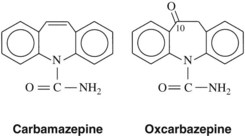
Phenobarbital, introduced in 1912, was the first drug used extensively to treat seizures. Between 1938 and 1960, numerous anticonvulsant agents were introduced, including the hydantoins, succinimides, and primidone. Between 1960 and 1992, several novel anticonvulsants were introduced (e.g., carbamazepine, valproic acid, clonazepam, clorazepate). With the passages of the Expedited Drug Approval Act and Prescription Drug User Fee Act in 1992, the approval process was facilitated, and 10 agents have since been introduced (with several more currently in clinical trials). Many of these drugs have been approved as adjunctive agents for use with earlier drugs in the treatment of “partial onset seizures”; these indications have broadened with increased experience in their use. In some cases the newer agents are referred to as second-generation and third-generation agents, and in several cases newer agents are related to older agents, such as phenytoin and fosphenytoin; carbamazepine and oxcarbazine; and meprobamate, felbamate, and fluorofelbamate (the last mentioned in premarketing trials).34
Drugs are described as having characteristic spectra for treating the various forms of seizures (Figure 14-1). Prescribing antiepileptic drugs for conditions outside their spectra may lead to problems beyond simple therapeutic failure. In particular, absence seizures can be exacerbated by many of the drugs used to treat tonic-clonic seizures. Some children “outgrow” absence epilepsy but have a tendency to develop other forms of epilepsy in later years. The discovery of valproic acid, which can control many forms of epilepsy, was a major breakthrough for patients in whom absence seizures convert to tonic-clonic seizures. The careful withdrawal of anticonvulsant therapy in children with a history of tonic-clonic epilepsy, but who have been seizure-free for several years, is sometimes successful. Finally, adults whose seizures were few in number before initiation of treatment and are well controlled with a single anticonvulsant may be weaned after 2 years of therapy with a reasonable expectation (>50%) of avoiding relapse.
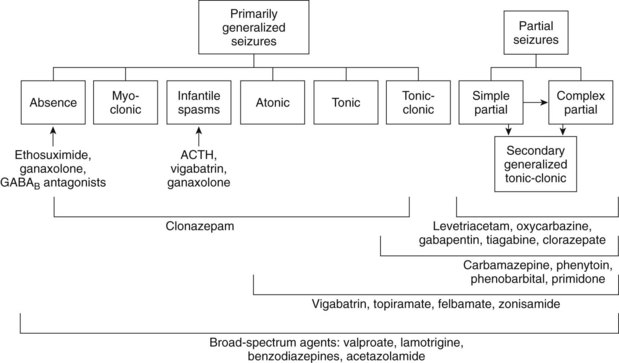
Different anticonvulsant drugs can be used for different aspects of seizure disorders. Anticonvulsant is a term that has used for agents that terminate seizures or status epilepticus events.17 Antiepileptogenic agents are used to prevent the development of epilepsy after a seizure-triggering event. The term anti-ictogenic refers to drugs that prevent the reoccurrence of seizures in an individual with a diagnosis of epilepsy. In this context, benzodiazepines are used as anticonvulsants for the emergency treatment of seizures in the dental office, although their use as anti-ictogenic drugs is limited by the development of tolerance to their anticonvulsant actions.
Typically about 50% of patients respond to traditional agents, and between 20% and 40% of the remainder respond to the addition of a supplemental agent. The drugs used to treat epilepsy and their proposed mechanisms of action and current indications are summarized in Table 14-2.
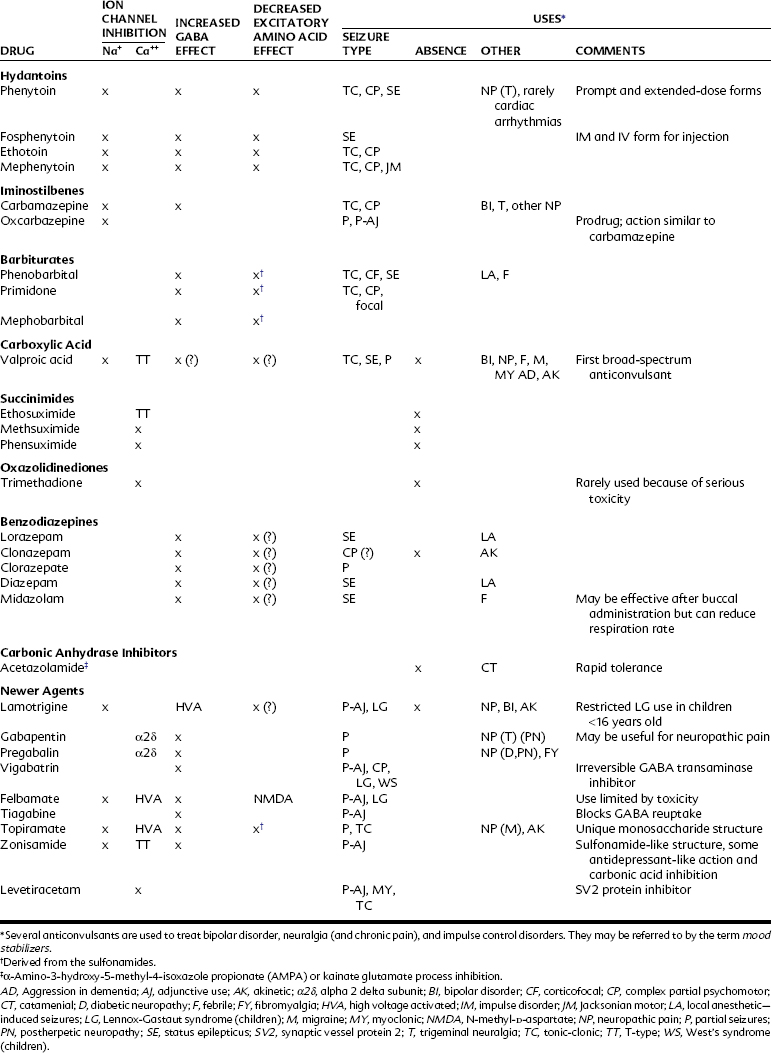
Several antiepileptic drugs can alter liver enzyme function. A cluster of adverse reactions and drug interactions can result from induction of hepatic enzymes, which may alter the metabolism of (1) the inducing anticonvulsant agent; (2) other drugs, altering their half-lives or toxicity; (3) vitamins (folate, vitamins D or K), which can produce vitamin deficiency disorders such as megaloblastic anemia, decreased bone density, fetal toxicity, or bleeding disorders; and (4) hormones (thyroid hormone or birth control pills). Drug effects on liver microsomal enzyme activity are summarized in Box 14-1. Carbamazepine, phenobarbital, phenytoin, and primidone are well-documented induction agents for the oxidative cytochrome P450 pathway and for phase II synthetic or conjugation elimination pathways (including uridine diphosphate glucuronosyltransferase [UGT]) and in some cases for P-glycoprotein or multidrug resistance proteins (MDR), which may play a role in multiple anticonvulsant drug resistance and poor seizure control. Phenobarbital, phenytoin, carbamazepine, felbamate, lamotrigine, gabapentin, and topiramate bind to P-glycoproteins that seem to facilitate their elimination from the brain. Lamotrigine selectively inhibits UGT. Valproate and topiramate may inhibit oxidative enzymes, prolonging the actions of other drugs. Oxcarbazepine and phenytoin may also inhibit some liver enzymes, as shown in Box 14-1.
BOX 14-1 Adverse Effect of Antiepileptic Drugs on Liver Microsomal Enzymes
Additional adverse reactions associated with anticonvulsant drugs include gingival overgrowth, aplastic anemia, hepatotoxicity, renal stones, visual disturbances, and fevers. These may represent pharmacogenomic processes or poorly understood aspects of their pharmacologic features in susceptible patients. Sometimes the reaction is manifested as teratogenicity or cancer; these delayed toxicities are dose independent, but host dependent. More recent studies have found new evidence that anticonvulsant drug use contributes to an increased incidence of birth defects.19 Behavioral, neurologic, and psychiatric reactions are common and can occur with several of the anticonvulsants. Drugs that facilitate GABA or inhibit glutamate pathways may be more likely to induce amnesia.
Anticonvulsant drugs can paradoxically promote seizure activity or precipitate new seizure types. Carbamazepine can increase absence and other seizures. Other anticonvulsants that may exacerbate seizures include phenytoin, phenobarbital, vigabatrin, oxycarbazine, lamotrigine, gabapentin, felbamate, and tiagabine.12 Increased seizure frequency is more likely in patients with severe seizure disorders.
Newer agents are expected to have more favorable safety profiles based on their different mechanisms of action and their lessened interaction with the microsomal drug metabolizing system. A full understanding of the clinical toxicology of drugs can take years to develop, however. In the case of vigabatrin, early reports about the drug can be found in the 1970s, but the first report of patients commonly (≥30%) developing irreversible visual field defects was published in 1997.15 Gabapentin was found to have a low side-effect profile in evaluation trials but is now being used at doses that are many times greater than were typically studied. Experts have noted that much of what is known about the new anticonvulsant drugs has been derived from manufacturer-sponsored trials. Differences in studied patient populations, dosages used, and the end points reported make clinically meaningful comparisons problematic; larger comparison studies by independent groups are still needed.9
CHEMISTRY AND STRUCTURE-ACTIVITY RELATIONSHIPS
Figure 14-2 shows the common structure present in all the clinically effective anticonvulsants developed before 1960. Substitution at position 1 of the ring results in the various classes of anticonvulsants indicated in Table 14-3.
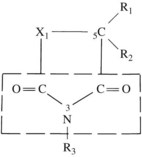
TABLE 14-3 Classes of Anticonvulsants According to Substitution at Position X1 of the Chemical Structure* (See Figure 14-2)
| ANTICONVULSANT | SUBSTITUTION |
|---|---|
| Barbiturates | –CO–NH– |
| Hydantoins | –NH– |
| Succinimides | –CH2– |
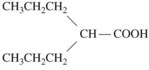
A phenyl ring at R1 or R2, such as appears in phenytoin, is a highly desirable, although not crucial substituent for protection against tonic-clonic epilepsy. An alkyl substituent at R1 or R2, such as appears in ethosuximide, is desirable (but not crucial) for control of absence seizures. A great deal of detailed structure activity information was obtained to identify opportunities for developing improved agents. More recent discoveries have included several agents with unrelated structures, however. Valproic acid is n-dipropylacetic acid (Figure 14-3), a simple branched-chain carboxylic acid, and carbamazepine (Figure 14-4) is chemically related to the tricyclic antidepressants and is used in the treatment of certain affective disorders (see Chapter 12).
HYDANTOINS
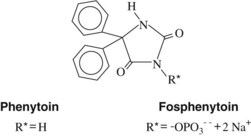
Phenytoin (diphenylhydantoin) is one of the first drugs to be discovered through an organized scientific search for a therapeutically effective compound. Introduced in 1938, phenytoin was immediately recognized as a breakthrough in anticonvulsant therapy because it suppressed seizures without causing as much sedative effect as phenobarbital. Phenytoin is an effective anticonvulsant against tonic-clonic and partial seizures and an important pharmacologic tool that has increased understanding of the underlying mechanisms responsible for epileptic syndromes. Mephenytoin and ethotoin are hydantoins related to phenytoin but are now rarely used. Fosphenytoin, the newest hydantoin, is a phosphorylated prodrug that is rapidly converted to phenytoin by endogenous phosphatase enzymes. It is water soluble and is better tolerated by parenteral administration. The structures of phenytoin and fosphenytoin are shown in Figure 14-5.
Pharmacologic Effects
The major site of action of phenytoin seems to be at the Na+ channel, and various actions have been shown at this site. The only mechanism evident at concentrations equivalent to therapeutic plasma concentrations (10 µg/mL to 20 µg/mL) is a reduction, however, in sustained high-frequency neuronal firing caused by phenytoin binding reversibly to inactivated Na+ channels.27 Phenytoin delays the neuronal recovery process whereby Na+ channels cycle from the refractory, inactivated state to the responsive, closed configuration, which is required before an action potential can be generated again. Phenytoin binding to inactivated Na+ channels is frequency and voltage dependent so that it becomes greater as neuronal depolarization and firing frequency increase. These properties are ideally suited for anticonvulsant activity because high-frequency neuronal discharge is characteristic of the epileptic disorders.
High extracellular K+, typically found during seizures, also increases the effectiveness of phenytoin. Normal (slower) neuronal activity is unaffected by phenytoin, which may explain its minimal sedative effects. At slightly greater than therapeutic concentrations, phenytoin interferes with Ca++ channels and the interaction of Ca++ and calmodulin, which disrupts Ca++-dependent phosphorylation of proteins necessary for neurotransmitter release from presynaptic nerve terminals. There are also some reports of phenytoin facilitating GABA or inhibiting glutamate processes.8 Phenytoin has also been found to alter the metabolism of some growth factors, which could play a role in neuroprotective actions of the drug (see later).
Absorption, Fate, and Excretion
Phenytoin given by intravenous injection can produce thrombophlebitis, arrhythmia, and hypotension. These side effects are largely caused by the vehicle needed to solubilize phenytoin for injection. Intramuscular injection of phenytoin may precipitate in the muscle, cause pain, and be poorly absorbed. Fosphenytoin is a water-soluble analogue that may be given intravenously or intramuscularly. After intramuscular administration, it produces much less pain and is absorbed rapidly.35
Adverse Effects
Phenytoin promotes gingival overgrowth in approximately 10% to 30% of all patients. Gingival overgrowth is usually more severe in children, for whom its incidence may be 50%. The primary mechanism responsible for this side effect is unknown. Several hypotheses have been proposed involving inflammation, bacterial plaque, the presence of teeth or dental implants, gingival fibroblast phenotype, epithelial growth factor, collagenase activation, folic acid deficiency, Na+/Ca++ flux, and perhaps salivary delivery of phenytoin into the mouth.2 It has been observed more recently that phenytoin increases platelet-derived growth factor B and its mRNA from macrophages that are thought to induce gingival fibroblast proliferation and local angiogenesis.20 The transforming growth factor-β pathway involving Grb1, SOS-RAS-ERK1/2, AP1, and Ca++ signaling pathways has been implicated in hereditary gingival overgrowth and may play a role in drug-induced gingival overgrowth.13 The result is an increase in fibroblast cell growth with increased interstitial ground substance.38 Other drugs that induce gingival overgrowth include the immunosuppressant cyclosporine and the dihydropyridine Ca++ channel blocking drugs. A more recent investigation has found that all of these drugs have the ability to reduce apoptosis (programmed cell death), suggesting that reduced cell loss rates could also play a role in gingival overgrowth.24 Figure 14-6 represents a possible model of gingival overgrowth.
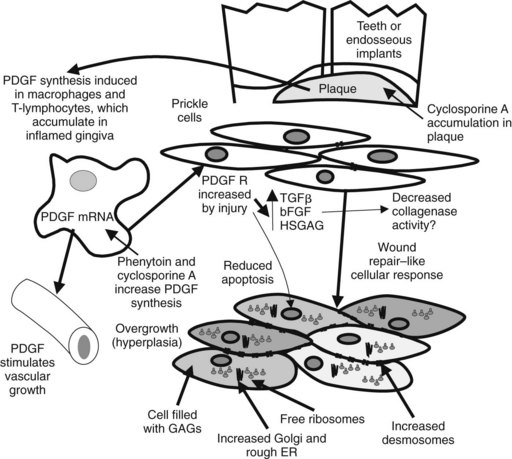
FIGURE 14-6 Effect of phenytoin and cyclosporine on gingival overgrowth. Predisposing factors include the presence of teeth or implants, inflammation, and overgrowth-inducing drugs. Phenytoin increases by sixfold platelet-derived growth factor (PDGF) mRNA in reparative/proliferative macrophages.38 PDGF is thought to increase angiogenesis and wound repair. Increases in fibroblastic growth factors, such as transforming growth factor-β (TGFβ) and basic fibroblast growth factor (bFGF), and production of heparin sulfate glycosaminoglycan (HSGAG) are induced by PDGF acting on its receptor (R). Prickle cells in the gingiva become filled with glycosaminoglycans (GAGs), rough endoplasmic reticulum (ER), and ribosomes, and their connective desmosomes proliferate (bottom).
Phenytoin may also cause numerous other side effects as summarized in Table 14-4. Phenytoin interferes with the metabolic activation of vitamins D and K, and the absorption of Ca++. Although the resultant effect on bone metabolism is usually subclinical, overt cases of rickets and osteomalacia have been observed.33 Vitamin D or K supplements may prevent these conditions.19,33 Vitamin K modulates the synthesis of osteocalcin and matrix G1a proteins, which influence Ca++ metabolism in bone. Children born to mothers who have received ph/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


