CHAPTER 13 Sedative-Hypnotics, Antianxiety Drugs, and Centrally Acting Muscle Relaxants*
The drugs discussed in this chapter have the common pharmacologic characteristic of being central nervous system (CNS) depressants, and they are capable of inducing various clinical responses, including relief of anxiety, sedative-hypnotic effects, and centrally acting muscle relaxation. Although all such drugs induce CNS impairment, drugs in certain categories have some degree of selectivity that determines their therapeutic indications in medical and dental practice. The ability of these agents to induce sedation, hypnosis, anxiolysis, or muscle relaxation selectively is limited, however, and significant overlap in the clinical indications for these drugs occurs. Pharmacokinetic differences and differences in mechanisms of action often distinguish these agents. The multiple actions and uses of these agents are also discussed in other chapters addressing anticonvulsants (see Chapter 14), general anesthetic agents (see Chapter 18), and antihistamines (see Chapter 22).
Insomnia is the salient feature of the nearly 90 different forms of sleep disorders.11 Epidemiologic studies report that insomnia is widespread, affecting one third of the population. Insomnia is more prevalent among women than men and is more common in elderly individuals than in younger individuals. Nearly half of all Americans older than 65 years experience sleep disorders.41
According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV),10 the anxiety disorders comprise various acute and chronic anxiety and phobic states. Specific anxiety disorders include panic disorder with or without agoraphobia, agoraphobia without panic disorder, generalized anxiety disorder, obsessive-compulsive disorder, acute stress disorder, post-traumatic stress disorder, social phobia, specific (simple) phobia, substance-induced anxiety disorder, and anxiety resulting from a general medical condition. The major emphasis in this chapter is on drugs effective against anxiety as a symptom rather than as a specific disorder. Although antianxiety drugs have applications for treatment of anxiety disorders in general, other drugs, including tricyclic antidepressants, monoamine oxidase inhibitors, and selective serotonin reuptake inhibitors, are used in the pharmacotherapy of panic disorders, phobic disorders, and obsessive-compulsive disorders. These latter agents are discussed in detail in Chapter 12.
BENZODIAZEPINES
Chemistry and Structure-Activity Relationships
The structures of the pharmacologically active 1,4-benzodiazepines are shown in Figure 13-1 and Table 13-1. All benzodiazepines currently available in the United States are derived from the basic molecule shown in Table 13-1, to which are added various substituent groups. Slight modifications of the basic structure have produced triazolobenzodiazepines (e.g., alprazolam, triazolam) and imidazobenzodiazepines (e.g., midazolam).
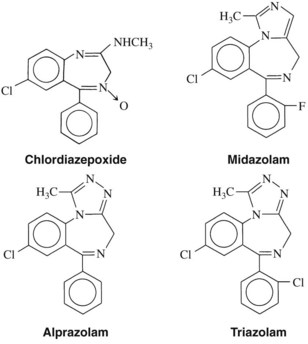
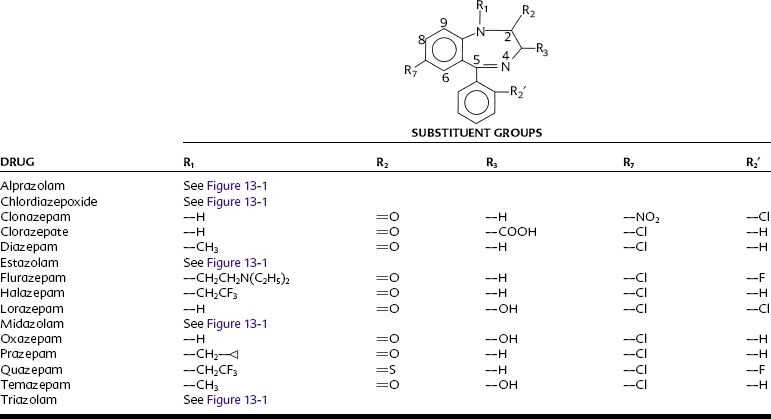
All benzodiazepines with psychopharmacologic activity have an electronegative group at R7. A chlorine atom seems to confer optimal activity, whereas bromo and nitro substitutions are only weakly anxiolytic. A nitro moiety at R7 enhances antiseizure properties, however, as illustrated by clonazepam, which is used as an anticonvulsant. Hydrogen or methyl groups at R7 significantly reduce pharmacologic activity. Substitution at position 5 with any group other than a phenyl ring also reduces activity. Halogenation at R2′ increases potency; larger alkyl substitutions decrease it. Substitution on the nitrogen at R1 with a methyl group enhances activity, as do methyl or hydrogen groups at R3. A biosynthetic pathway for the in vivo formation of diazepam-like benzodiazepines has been proposed.7 Whether synthesis occurs naturally is unknown, but benzodiazepines are found in a variety of foods.69
Mechanism of Action
Perhaps the most exciting and significant advance in the understanding of anxiety and the mechanism of action of benzodiazepines occurred with the discovery of specific benzodiazepine binding sites in the brain and the understanding that these were in some way linked to the inhibitory neurotransmitter γ-aminobutyric acid (GABA). As shown schematically in Figure 13-2, when the GABA receptor is activated, the Cl− channel opens, allowing Cl− influx, membrane hyperpolarization, and neuronal inhibition. Benzodiazepines, by interacting at high-affinity benzodiazepine binding sites on the GABA receptor complex, facilitate GABA action. Although devoid of direct GABA-mimetic effects, benzodiazepines increase inhibitory neurotransmission resulting from GABA. Although the exact mechanism by which benzodiazepines accomplish their effect is not fully delineated, it is known that they increase the frequency at which Cl− channels open in response to GABA.60 GABA inhibition (chiefly postsynaptic inhibition) is enhanced by benzodiazepines, and any transmitter system modulated by this inhibitory drive is inhibited to a greater extent in the presence of benzodiazepines.
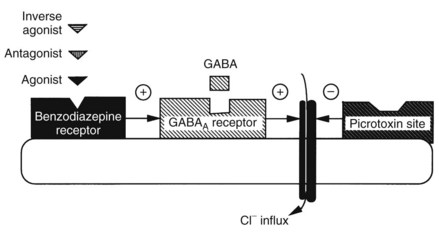
Benzodiazepine receptors are found in the brains of all mammalian species, birds, amphibians, reptiles, and higher fishes. Benzodiazepine receptors are linked to a specific GABA receptor subtype, the GABAA receptor (see Figure 13-2). Figure 13-3 provides further details on binding domains associated with the GABAA receptor. Historically, GABA receptors have been classified into two subtypes: the Cl− channel–linked GABAA receptors and the G protein–linked GABAB receptors. Benzodiazepine-sensitive GABAA receptors are activated by GABA agonists, such as muscimol (a hallucinogen), and blocked by GABA antagonists, such as picrotoxin and bicuculline (convulsants).72 GABAB receptors are benzodiazepine and bicuculline insensitive and are activated by baclofen, a centrally acting muscle relaxant.
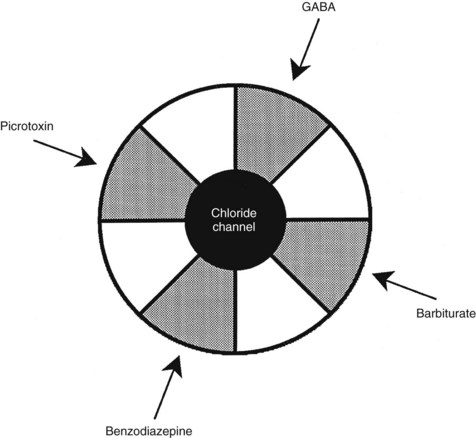
The benzodiazepine receptor—along with the GABAA receptor, a barbiturate receptor, the Cl− channel, and binding domains for other drugs—forms a single macromolecular complex. Similar to GABA receptors, benzodiazepine receptors are heterogeneous; there are at least three types: type 1 (BZ1), type 2 (BZ2), and the “peripheral type” benzodiazepine receptor. The presence of BZ1 and BZ2 receptor types is apparently determined by the subunit composition of the GABAA macromolecular complex. The BZ1 receptor may be linked to sleep, whereas the BZ2 receptor may be linked to cognition and motor function. High-affinity benzodiazepine binding sites are found on specific subunits of the GABAA receptor complex, which, as shown in Figure 13-4, is a pentamer composed of several glycoprotein subunits (α, β, γ). This organization is analogous to the organization of the nicotinic receptor. As illustrated in Figure 13-4, which depicts the most common form of GABAA receptor complex in the rat brain, a γ subunit is necessary (but insufficient) for benzodiazepine binding and pharmacologic effects.56 Cloning experiments have shown that there are multiple subtypes of α, β, and γ subunits,33 which provide a basis for GABA receptor heterogeneity.13,49
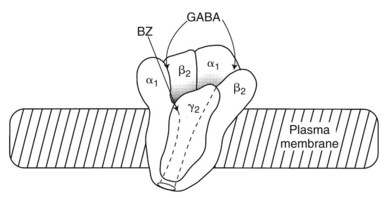
The heterogeneity of receptor subunits may offer an explanation for the diverse pharmacologic effects (antianxiety, anticonvulsant, sedative, and skeletal muscle relaxant) of benzodiazepines. Determination of the molecular basis of receptor heterogeneity may eventually facilitate the development of benzodiazepines with a greater degree of selectivity in producing each of these effects. At present, none of the clinically available antianxiety benzodiazepines shows selectivity for either BZ1 or BZ2 receptors, although the hypnotic benzodiazepine quazepam is likely selective for the BZ1 receptor.50 Zolpidem and zaleplon, two nonbenzodiazepines selective for the BZ1 receptor, are discussed later in this chapter.
The benzodiazepine-insensitive GABAB receptors coupled to G proteins are associated with a decrease in Ca++ conductance and an increase in K+ conductance and could be expected to cause pharmacologic effects when stimulated or antagonized. GABAB receptors are less widely distributed than GABAA receptors but are found in high concentrations in the cerebral cortex and cerebellum. Subtypes of GABAB receptors may exist. GABAB receptors have not been studied as extensively as GABAA receptors, but they may participate in blood pressure regulation61 in addition to muscle activity and offer a potential site for therapeutic drug action.
The existence of subclasses of benzodiazepine receptors suggests that some agents, with specific activity for individual receptor subtypes, may be more selective than others in terms of their pharmacologic profile. Whether this selectivity results in significant clinical differences is an open question.14 Quazepam, a long-acting benzodiazepine hypnotic, produces sedation, but seems to have little ataxic effect and may cause less tolerance than other benzodiazepines. Autoradiographic studies have shown selective binding of quazepam to BZ1 receptors,31 which may account for sedation with minimal muscle relaxant effects. Of all currently available benzodiazepines, only quazepam, one of its active metabolites (1-oxoquazepam), and possibly the antianxiety agent halazepam have selectivity for the BZ1 receptor subtype. These benzodiazepines differ chemically from other benzodiazepines by having a trifluoroethyl substituent (see Table 13-1), which may be responsible for BZ1 selectivity. Selective activity at the BZ1 receptor has not been associated with any special clinical benefit of quazepam, however, compared with other benzodiazepines for treating insomnia.
Although the pharmacologic actions of benzodiazepines are closely tied to GABA receptors, numerous other neurotransmitters, including glycine, norepinephrine, and 5-hydroxytryptamine (5-HT), have been suggested to play a role in their action. An interaction between GABA and 5-HT has been shown experimentally with diazepam and tryptaminergic anxiolytics.36 This finding is interesting in light of the mechanism of action of the nonsedating antianxiety agent buspirone (see later), a 5-HT1A partial agonist.
Pharmacologic Effects
Benzodiazepines have clinically useful antianxiety, sedative-hypnotic, amnestic, anticonvulsant, and skeletal muscle relaxant properties. Benzodiazepines previously were thought to differ pharmacologically only in terms of their pharmacokinetics. Although differences in pharmacokinetic properties explain many of their clinical differences, certain benzodiazepines seem to have unique properties.17 Alprazolam has documented antidepressant and antipanic properties, and diazepam may be more selective as a skeletal muscle relaxant than other benzodiazepines. Diazepam is the only benzodiazepine approved for the treatment of skeletal muscle spasm and spasticity of CNS origin.
Central nervous system
Certain benzodiazepines in clinical doses can induce anterograde amnesia, which means that memory of events occurring for a time after drug administration is not retained.12 This effect is useful therapeutically in intravenous sedation or monitored anesthesia care. Muscle relaxation and antiseizure activity are additional CNS effects of benzodiazepines. These effects are discussed later in this chapter (muscle relaxation) and in Chapter 14 (antiseizure activity).
Respiratory system
As is true of any sedative drug, benzodiazepines are respiratory depressants. In normal doses, benzodiazepines have little effect on respiration in healthy individuals. There have been reports, however, of benzodiazepine-induced respiratory failure in patients with pulmonary disease. Benzodiazepines may cause additive respiratory depressant effects with other CNS depressant drugs. Poor suckling, hypothermia, and a need for ventilatory assistance have been reported in neonates of mothers who received intravenous lorazepam shortly before delivery. Midazolam, used primarily for intravenous sedation and for the induction of anesthesia, can cause respiratory depression and apnea. Clinically significant respiratory depression may occur if an opioid is used in combination with midazolam.12
Absorption, Fate, and Excretion
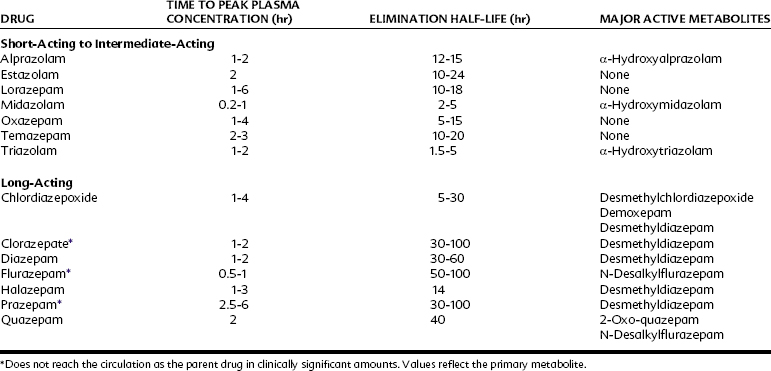
The pharmacokinetics of individual benzodiazepines differ, and there is a wide range in speed of onset and duration of action among these compounds. Benzodiazepines frequently are classified according to their elimination half-life, as illustrated in Table 13-2; however, the elimination half-life of a given drug is only one factor affecting its clinical profile. The rates of drug absorption and tissue distribution and redistribution are often important factors in determining onset and duration of clinical effects after short-term administration. Additionally, there is a wide variation in drug half-lives among patients.
TABLE 13-2 Classification of Benzodiazepines on the Basis of Elimination Half-Life after Oral Administration
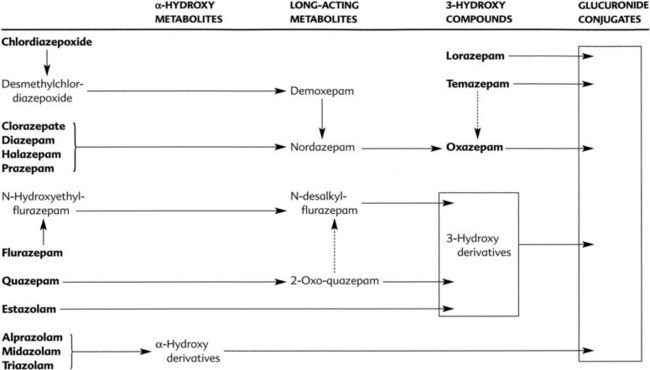
Many benzodiazepines are converted to pharmacologically active metabolites that have long half-lives (Figure 13-5). Clorazepate and prazepam are nearly completely converted (in the stomach and liver) to the long-acting metabolite desmethyldiazepam (nordazepam) before they enter the systemic circulation. Desmethyldiazepam is a metabolite of many other benzodiazepines, including chlordiazepoxide, diazepam, and halazepam. Flurazepam is also converted to active metabolites in its first pass through the liver. Generally, the products of phase I metabolism are eventually conjugated with glucuronic acid and inactivated and excreted in the urine and feces. Because the half-lives of the different active metabolites vary considerably, the overall duration of the pharmacologic effect of benzodiazepines also varies considerably. Oxazepam and lorazepam are not converted to active metabolites but are directly conjugated and excreted. These drugs are eliminated rapidly and may be especially useful in patients who have a deficiency in hepatic microsomal enzymes resulting from liver disease or other reasons.
The poor oral bioavailability of triazolam, alprazolam, and midazolam of approximately 50% is believed to be due to CYP3A4 metabolism in the gut wall and hepatic first-pass metabolism. Triazolam’s availability is improved when administered sublinqually.32 Inhibition of CYP3A4 metabolism by coadministration of itraconazole, erythromycin, or grapefruit juice can significantly increase maximum blood concentrations and the area under the curves of these short-acting benzodiazepines.27
Adverse Effects and Drug Interactions
Tolerance and psychological dependence develop frequently with benzodiazepines, but true physical dependence is less common. Nevertheless, the abuse potential of benzodiazepines should not be ignored.51 Tolerance to the sedative-hypnotic effects of benzodiazepines is slower to develop with longer acting agents. In cases of physical dependence, the severity of withdrawal depends on the dose of the drug being used and the drug’s half-life. Rapid discontinuation of benzodiazepines, especially short-acting compounds, can lead to symptoms of withdrawal. Often these symptoms are nearly identical to the symptoms for which treatment was initiated, including anxiety, irritability, insomnia, and fatigue. The symptoms become more severe with high doses and prolonged treatment. Withdrawal can be minimized by reducing the dosage very gradually (≤10% per day over 10 to 14 days) or by the use of longer acting compounds. Withdrawal from lower doses is usually not life-threatening, and symptoms last no longer than 2 weeks. Withdrawal from high doses may be life-threatening because of accompanying convulsions.
Mechanisms involved in the development of tolerance are unknown, but the long-term administration of benzodiazepines to animals causes downregulation of benzodiazepine receptors,40 which could be a contributing factor. Diazepam has been particularly popular as a drug of abuse. Because of the strong binding of diazepam to tissue constituents, it is not rapidly removed by dialysis or diuresis in patients with acute overdose. Flumazenil, a benzodiazepine antagonist (described later), can reverse benzodiazepine overdose. Flumazenil can precipitate withdrawal in benzodiazepine-dependent patients, however.
Benzodiazepines cross the placental barrier. During the first trimester, long-term use of these drugs has been associated with increased fetal malformations, including cleft lip and cleft palate in humans. There is no clear estimate of the risk after single-dose use. All benzodiazepines are classified as pregnancy category D except triazolam, which is pregnancy category X. It is generally agreed that these drugs should be avoided during pregnancy.43 The frequent use of benzodiazepines during late pregnancy may lead to withdrawal in the neonate. Large doses of benzodiazepines given to mothers during labor and delivery may result in respiratory depression, hypotonia, and hypothermia in neonates.
Drug interactions associated with anxiolytic and sedative drugs used in dentistry are listed in Table 13-3. The therapeutic index for benzodiazepines is normally so large that wide ranges of dosing recommendations and blood concentrations do not significantly affect their safety and efficacy. Plasma concentrations after a given dose may normally vary such that a minor shift in elimination from drug interactions is unlikely to result in an overdose. In healthy subjects taking no other medications, plasma concentrations 3 hours after a single 15 mg dose of diazepam have been reported to range from 20 µg/mL to 260 µg/mL.38 A drug interaction that causes a 20% increase in diazepam plasma concentrations is unlikely to have significant toxicity. Most healthy patients can tolerate small variations in a drug’s absorption or metabolism that are caused by coadministration of another drug. Combining sedatives is problematic. The combination of ethanol with a benzodiazepine is an important source of serious toxicity.63
TABLE 13-3 Adverse Drug Interactions: Anxiolytics and Sedative-Hypnotics
| ADVERSE DRUG INTERACTION (SPECIFIC EXAMPLES) | CLINICAL IMPLICATIONS |
|---|---|
| Anxiolytics and Sedative-Hypnotics with: | |
| Other anxiolytics and sedative-hypnotics, alcohol, opioids, antipsychotics, antidepressants, centrally acting muscle relaxants, local and general anesthetics, and other CNS depressants | In combination, CNS depression summates with anxiolytics and sedatives; loss of consciousness, respiratory depression, and death are possible complications |
| Benzodiazepines with: | |
| Carbamazepine, rifampin | Increased rate of metabolism reduces bioavailability of several benzodiazepines |
| Cimetidine, diltiazem, verapamil, erythromycin, clarithromycin, protease inhibitors (indinavir, nelfinavir, ritonavir), some azole antimycotics (itraconazole, ketoconazole), and some antidepressants (fluoxetine, fluvoxamine, trazodone) | Decreased rate of metabolism increases bioavailability of some benzodiazepines and significantly augments and prolongs their effects |
| Chloral Hydrate with: | |
| Alcohol | Each drug limits metabolism of the other; depression is greater than additive |
| Warfarin | Competition for plasma protein binding causes temporary increase in anticoagulant effect |
| Furosemide | Rare reports of diaphoresis, tachycardia, and hypertension |
| Epinephrine | Myocardial sensitization and cardiac dysrhythmias |
| Barbiturates with: | |
| Valproic acid and phenobarbital | Elimination of barbiturates is decreased; prolonged and enhanced sedation is reported |
| Warfarin | Bleeding risk increases when long-term barbiturate therapy is discontinued |
| Anticoagulant effect of warfarin is reduced with concurrent therapy with phenobarbital |
CNS, Central nervous system.
Rifampin induces metabolic enzymes in the gut and liver responsible for the metabolism of diazepam, midazolam, and triazolam. A 96% reduction in the bioavailability of midazolam has been reported.2 Triazolam is so rapidly and effectively metabolized in the gut that peak plasma concentrations are only 12% of normal.68 This interaction is one of the most pronounced alterations in drug kinetics ever reported. The almost complete loss of triazolam bioavailability and subsequent efficacy is quite significant and warrants use of an alternative anxiolytic, such as oral oxazepam, nitrous oxide inhalation, or an intravenous agent. The anticonvulsant carbamazepine can also induce hepatic enzymes for the oxidative metabolism of benzodiazepines such as alprazolam, triazolam, and midazolam.3 Decreased benzodiazepine plasma concentrations and greatly reduced sedative effects after oral administration of these agents may occur. This interaction may be important in medicine because of loss of seizure control. A loss of sedative efficacy in dentistry may also occur. Benzodiazepines that are metabolized solely through glucuronidation, such as oxazepam, are suitable alternative agents for sedation in these situations.
The Ca++ channel blockers verapamil and diltiazem have been shown to inhibit the CYP3A isozymes required for the metabolism of triazolam and midazolam. In controlled clinical trials, a 2-day regimen of these drugs decreased the metabolism and increased the bioavailability of midazolam and triazolam administered orally. Peak blood concentrations were increased twofold to threefold and were associated with increased sedation and performance deficits.1 Avoidance of this combination is recommended, particularly in elderly patients known to be sensitive to benzodiazepines.
Cimetidine also inhibits the oxidative metabolism of certain benzodiazepines, such as triazolam and alprazolam. Half-life increases of 30% to 63% have been reported.18 Metabolism of diazepam may also be delayed. An increased and prolonged level of sedation after oral administration may occur because of the decreased first-pass metabolism.52 Benzodiazepines that are metabolized directly to the glucuronide conjugate (e.g., oxazepam) are not affected. Similarly, the antimicrobials erythromycin and clarithromycin and the azole antifungals ketoconazole and itraconazole are potential inhibitors of the hepatic isozymes required for oxidative metabolism of these benzodiazepines. By decreasing the first-pass effect and improving bioavailability, triazolam blood concentrations may increase threefold.67 The antiviral agents indinavir, nelfinavir, and ritonavir inhibit hepatic oxidative enzymes required for metabolism of many benzodiazepines. These significant pharmacokinetic drug interactions could potentially cause oversedation and respiratory depression.
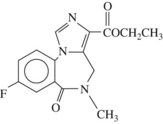
Antagonists
Benzodiazepine antagonists are important therapeutic compounds that have the potential to reverse the effects of benzodiazepines. They have no intrinsic activity of their own but do not reverse constitutive benzodiazepine receptor activity. Flumazenil (Figure 13-6) is currently the only benzodiazepine receptor antagonist approved by the FDA.
Flumazenil has clinical application in managing benzodiazepine overdose and in hastening recovery from benzodiazepine sedation or anesthesia after diagnostic procedures or minor surgery. Although certain precautions must be observed, flumazenil may allow for a shorter monitoring period after surgery and earlier discharge of the patient.20 Flumazenil has been used successfully in reversing benzodiazepine-induced coma, but whether it should be given routinely to comatose patients when the cause of the coma is unknown is unclear. The routine use of flumazenil is not recommended in cases of mixed drug overdose, airway obstruction, or seizure disorders. Flumazenil may increase the risk of cardiac arrhythmias and seizures in patients who have overdosed with tricyclic antidepressants.22 Ventricular arrhythmias have also been precipitated by flumazenil in patients with chloral hydrate overdose.55
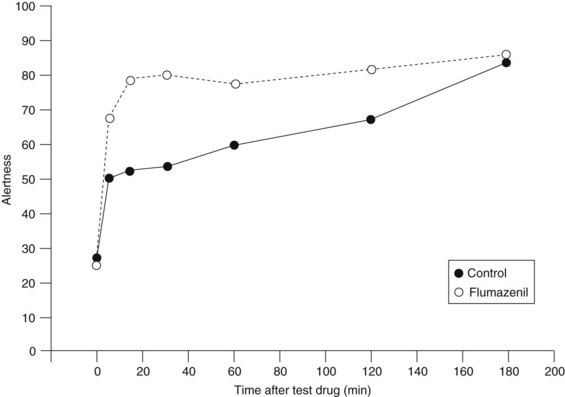
Flumazenil administered intravenously can generally reverse benzodiazepine-induced sedation in 1 to 2 minutes. Reversal of benzodiazepine sedation by flumazenil may last for several hours. In a study in which patients were sedated with midazolam before dental extraction, flumazenil significantly improved patient assessment regarding state of alertness compared with placebo controls (Figure 13-7) only for the first 30 minutes.8 The duration of action of flumazenil (elimination half-life of 45 to 75 minutes) is likely to be shorter than that of a benzodiazepine agonist. Other studies have also noted that the duration of action of flumazenil is shorter than that of midazolam and that sedation and respiratory depression may recur.20 Flumazenil is not a substitute for careful postoperative monitoring.
Another cautionary note is the possibility of flumazenil precipitating withdrawal in patients who are dependent on the benzodiazepines. Signs of benzodiazepine withdrawal include flushes, agitation, tremor, and seizures. Resedation with a benzodiazepine or barbiturate may be required in these circumstances. Although some studies suggest the amnesia from benzodiazepines is reversed by flumazenil, this is not consistently observed.9
General Therapeutic Uses
Approximately 35% of patients with a generalized anxiety disorder show marked improvement with benzodiazepines, 40% are moderately improved, and 25% remain unresponsive.15 These antianxiety agents are useful in the treatment of acute anxiety resulting from transient stress that is environmental, physical, or psychological in origin. For the treatment of long-standing anxiety, benzodiazepines ideally should be used only with appropriate psychotherapy. Sometimes benzodiazepines are prescribed inappropriately and with too little supervision. Despite concerns about the abuse potential of benzodiazepines, patients who have no prior history of drug abuse are unlikely to be at risk. Table 13-4 lists benzodiazepines and other drugs used for the management of acute anxiety.
| DRUG | USUAL DOSE* (mg) | ROUTE OF ADMINISTRATION |
|---|---|---|
| Alprazolam | 0.75-4.0 (adult) | Oral |
| 0.5-0.75 (elderly) | Oral | |
| Clorazepate | 15-60 (adult) | Oral |
| 7.5-15 (elderly) | Oral | |
| Chlordiazepoxide | 15-100 (adult) | Oral |
| 50-100 (adult) | IM, IV | |
| 10-20 (elderly) | Oral | |
| Diazepam | 4-40 (adult) | Oral |
| 2-20 (adult) | IM, IV | |
| 2-5 (elderly) | Oral | |
| 0.3-0.6 mg/kg (children) | Oral | |
| Halazepam | 60-160 (adult) | Oral |
| 20-40 (elderly) | Oral | |
| Lorazepam | 1.5-10 (adult) | Oral |
| 1-2 (elderly) | Oral | |
| 2-4 (adult) | IM, IV | |
| Midazolam | 2-10 (adult) | IM, IV |
| 0.25-1 mg/kg up to 20 mg (children) | Oral | |
| Oxazepam | 30-120 (adult) | Oral |
| 30-60 (elderly) | Oral | |
| Prazepam | 20-60 (adult) | Oral |
| 10-15 (elderly) | Oral | |
| Triazolam | 0.25-0/> |
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


