Metabolic Response to Trauma
Injury produces profound systemic effects. Hormones, the autonomic nervous system, and cytokines all produce a series of responses that are teleologically designed to help defend the body against the insult of trauma and promote healing. Classically, these responses have been described as the stress response, a term coined by the Scottish chemist Cuthbertson in 1932.1 However, some of these responses may be counterproductive. The cascade of interactions is orchestrated in the severely traumatized patient to produce a host of responses that follow a recognizable pattern, but the depth and duration of these changes are variable, usually proportional to the extent of the injury and the presence of ongoing stimulation. Stresses other than major trauma produce alterations in the metabolic responses; examples are burns, sepsis, and starvation. Each results in marked variations in the metabolic response, and this variability persists during the later chronic and recovery phases of the original injury (Fig. 1-1).
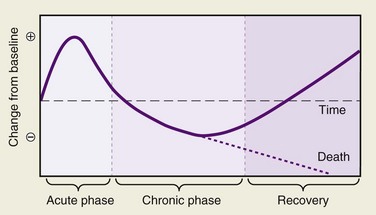
The body’s initial response to insult (the acute phase) is directed at maintaining adequate substrate delivery to the vital organs, in particular oxygen and energy. Cuthbertson’s pioneering work recognized the increases in basal temperature, energy expenditure, and oxygen consumption, and also the loss of potassium and nitrogen.2,3 The term systemic inflammatory response syndrome (SIRS) is used to describe the body’s response to infectious and noninfectious causes and consists of two or more of the following—hyperthermia or hypothermia, leukocytosis or leukopenia, tachycardia, and tachypnea. When the inflammatory response impairs function of organs or organ systems, the term multiple organ dysfunction syndrome (MODS) is used. As greater sophistication in the care of the multiply injured patient has permitted careful observation and analysis of the metabolic changes that accompany trauma; similar advances in the field of molecular biology have allowed the identification and measurement of the precise hormones and inflammatory mediators involved in the body’s response to trauma. This chapter reviews the mechanisms and consequences of the metabolic response to traumatic injury and some common approaches to the problems produced by these metabolic derangements.
Physiologic Response
The blood glucose level is also elevated through a complex and tissue-specific insulin resistance pathway.4 The maintenance of blood glucose levels supports the obligate glucose-metabolizing tissues—brain, bone marrow, erythrocytes, granulation tissue, and the immune system. Free fatty acids are mobilized by the catabolic hormones and become the main source of energy for peripheral tissue. Stored hepatic glycogen is limited and may be depleted within 1 day.
As the catabolic or flow phase is established, the metabolic emphasis is on the provision of substrate for healing. The basal metabolic rate is elevated, and thereafter the patient catabolizes muscle to make glucose. Birkhahn and colleagues found an almost 80% rise in catabolism after skeletal trauma, and Kien and associates have reported elevations as high as 103% in burn patients.5,6 The total body nitrogen balance becomes negative. Some of the earlier changes of the ebb phase may be reversed; for example, a diuresis may occur, or the heart rate may slow.
It may be difficult to separate the effects of the metabolic response to trauma from the effects of shock or incomplete resuscitation, but clearly such a distinction is necessary. The initial phase of the trauma response is complicated by an ongoing deficiency in tissue perfusion. The clinician must respond to dynamic changes in physiology to make time-sensitive interventions. The pulmonary artery or Swan-Ganz catheter offers a continuous window into the minute to minute mechanics of the severely injured patient. More recently, the safety and usefulness of Swan-Ganz catheters in trauma resuscitation has been scrutinized and their routine use has declined.7 Less invasive modalities such as the central venous catheter, near-infrared spectroscopy (NIRS), and continuous cardiac output monitoring by arterial waveform analysis are as effective at guiding therapy.8–10
The measured serum lactate level may help guide therapy and additional resuscitation. During shock states, tissue perfusion is decreased and increasing numbers of cells depend on anaerobic metabolism for energy. Normalization of serum lactate levels can indicate a return to global aerobic metabolic states and serve as an end point for resuscitation. Partly because of the reversible nature of hypoperfusion in the trauma setting, initial serum lactate levels are not themselves independent predictors of morbidity and mortality.11
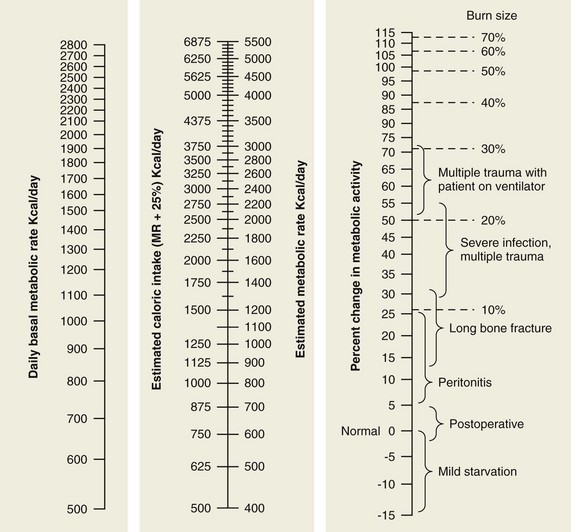
Similar injuries to different body regions can produce dramatic differences in metabolic requirement (Fig. 1-2). Two injuries in particular are worth special mention because of their notable metabolic consequences. The first is injury caused by head trauma. When brain injury is superimposed on multisystem trauma, the risk of death more than doubles.12 Increased mortality may not be directly caused by neurologic injury per se, but rather by the systemic consequences of deranged metabolism.13 In addition, many methods whereby brain-injured patients are managed after injury have themselves profound effects on metabolism. Examples of these interventions include steroids, systemic dehydration, muscle paralysis, hyperventilation, hypothermia, hyperbaric oxygen therapy, and barbiturate coma. The effectiveness of these treatments has been challenged but they continue to remain in use.14–16
Mediators of the Response
Neuroendocrine Response
An early response of the neuroendocrine system is the upregulation of the sympathoadrenal axis, which generates high plasma concentrations of epinephrine, norepinephrine, vasopressin, and dopamine. The peak and duration of the response parallel the severity of the trauma.17 These substances directly affect blood glucose levels and also produce inhibition of glucose uptake by tissue, which stimulates glucagon secretion. Sympathetic activity promotes lipolysis within adipose tissue, which begins to provide an energy source for gluconeogenesis.18 Gluconeogenesis in the liver is stimulated by glucagon.
The hypothalamic-pituitary axis is stimulated at the same time as the sympathetic nervous system after trauma.19 The pituitary releases adrenocorticotropic hormone (corticotropin, or ACTH), which stimulates the adrenal cortex to secrete glucocorticoid hormones such as cortisol and aldosterone. Epinephrine and cortisol promote muscle breakdown, protein catabolism, and amino acid release.
The effects of the flow phase of the metabolic response to trauma are partly attributable to hormones such as glucagon and cortisol, but not entirely, because the catabolic consequences extend beyond measurable elevated levels of these hormones.20,21 This finding has implicated other factors such as cytokines or the suppression of other hormonal axes such as those of somatostatin and growth hormone.
Cytokines
Thought to be the most proximal mediator of the inflammatory response, TNF was originally described as the catabolic factor cachectin.22 At least two forms of TNF exist.23,24 TNF influences cellular attraction as part of the local inflammatory response, leukocyte migration, and systemic hypotension.25,26 It also promotes muscle catabolism, free fatty acid release, and hepatic synthesis of acute-phase reactants. TNF free receptor is a glycosylated protein found in membrane-bound and free-floating forms. In contrast to IL-1, TNF appears to act peripherally and has no direct effect on lymphocyte activation.27
The interleukins are polypeptides released from lymphocytes; each is numbered according to the amino acid sequence that elicits its action.28 Circulating free receptors are known for IL-1 and IL-6. Free receptors may exist for all cytokines. They appear to function in the regulation of cytokine activity. IL-1, which can be detected in the circulation within a few hours after injury, has multiple biologic effects, including the activation of resting T lymphocytes and macrophages, induction of hematopoietic growth factors, stimulation of chemotaxis of neutrophils, and synthesis of collagen and collagenases. More profound systemic effects include fever and changes in protein metabolism. Originally described as pyrexin, IL-1 was shown in subsequent studies to act via the production of prostaglandins in the hypothalamus and to alter the set point of the thermoregulator in the hypothalamus.29,30 It is in this way that antiprostaglandin agents, such as aspirin, can block the fever effect of IL-1.31 A byproduct of IL-1 metabolism can increase proteolysis of human muscle and induce hepatic protein synthesis.32,33 Identified as a B cell differentiation factor, IL-6 is now recognized as the final common mediator in a cascade of cytokine activity that alters hepatocyte protein synthesis.34,35
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


