Mechanical loading of bone is essential for maintaining bone mass and integrity. Conceptually, bone adapts to natural (weight bearing, muscle pull) and therapeutic (orthodontic) mechanical strains to achieve a better balance between mechanical stress and the load-bearing capacity of the bone tissue. For example, increased loading, as seen in the arms of tennis players, results in increased bone formation. In contrast, loss of loading, as during immobilization or spaceflight, can decrease bone formation and increase bone resorption, resulting in bone loss.
When an external force is applied to a bone, it results in displacement of particles from their original positions. Displacement differs from one particle to the next and results in deformation of shape or volume of the bone. This deformation is called strain. In a simple one-dimensional linear system, such as the uniform stretching of a wire, strain is defined as the fractional change in the length, ϵ = (change in length)/(original length). Although strain is dimensionless, it is common to measure it in microstrain (µϵ), or 10 −6 mm/mm. For example, a strain of 0.01 mm/mm would be equivalent to 10,000 µϵ, or 1%, which would be considered a large strain in bone. These displacements generate stresses equivalent to force per unit area (σ) at internal surfaces within the bone. For linear elastic solids, stress and strain are proportional, σ = (constant) × (ϵ).
For alveolar bone tissue subjected to mechanical loading, orthodontic forces must be converted into intracellular signals in mechanosensitive cells. This information must then be communicated to other nonmechanosensitive cells to produce a coordinated response. For this to occur, the following events must take place:
- 1.
External orthodontic forces must be converted into a signal detectable by the cell (transduction mechanism).
- 2.
The periodontal ligament (PDL) and alveolar bone must have cells that are able to detect mechanical loading–induced signals (mechanosensitive cells).
- 3.
Mechanosensitive cells must have a mechanism to sense the signal (mechanoreceptor).
- 4.
Mechanoreceptors must transduce loading information to intracellular signals.
- 5.
Intracellular signals within mechanosensitive cells must lead to the production and release of cellular mediators to communicate mechanical loading information to other cells.
Major responses of mechanosensitive bone and PDL cells to mechanical loading include activation of signaling pathways and new gene transcription, leading to the production of cellular mediators, such as nitric oxide (NO) and prostaglandin E 2 (PGE 2 ), which are thought to play a role in the local regulation of bone formation and resorption seen in orthodontic tooth movement.
MECHANOTRANSDUCTION
Transduction Mechanisms
When an orthodontic force is placed on teeth, it must be transduced into a signal detectable by mechanosensitive cells. Three possible transduction mechanisms have been proposed ( Fig. 26-1 ). First, loading of the periodontal ligament and bone causes deformation of the matrix. The mechanosensitivity of PDL cells, osteoblasts, and osteocytes is related to the amount of deformation or strain they experience.
Second, it has been proposed that cells themselves do not undergo significant deformation but are responsive to fluid shear stress generated by the deformation of matrix. Bone has been described as a “water-soaked sponge,” such that a compressive force on one side drives interstitial fluid toward the other side. The velocity with which the fluid flows is related to the rate at which the force is applied. This fluid flow through the canalicular-lacunar network creates shear stress on the surface of the osteocytes and bone-lining cells.
Third, it has been proposed that stress-generated potentials are responsible for the effects of mechanical loading on bone. The fluid in bone contains various ions. Movement of the ions by mechanical loads creates a stress-generated potential, and it has been shown that bone cells are responsive to electric fields. Current research indicates, though, that the actual changes in potential difference produced by streaming potentials are small compared with the electric potential difference induced by muscle contractions. The electric potential difference from the muscles completely overwhelms the local potential difference at the bone surface. Because the stimulus to bone remodeling from loading is usually associated with muscular activity, the effect of streaming potentials appears to be of minor importance in the mechanical loading–induced cell signaling in bone.
Whether the transduction mechanism is primarily caused by fluid flow or the actual deformation of the cell by the orthodontic force is still unknown. In the PDL, mechanical loading by the application of compression or tension has been shown to cause the upregulation of a variety of genes. In addition, pulsating-fluid shear stress of 0.6 MPa has been shown to cause upregulation of interleukin-8 (IL-8) gene expression, NO, and prostaglandin production, but whether orthodontic tooth movement produces this level of fluid shear stress in the PDL is debatable.
Evidence that strain by itself is not the mechanotransducer in bone cells comes from in vitro experiments. In one study, osteoblastic cells were incubated on polystyrene film and subjected to unidirectional linear strains by stretching of the film in the range of 500 to 5000 µϵ. There was no increase in the production of two factors thought to be important in mediating loading effects on bone, NO and PGE 2 , after loading. In contrast, the investigators found that exposure of osteoblastic cells to increased fluid flow induced both PGE 2 and NO production. Another study used a technique that produces uniform levels of strain and fluid shear stress and that permitted both shear stress and strain to be varied independently. Osteopontin (OPN) messenger ribonucleic acid (mRNA) expression, a marker of osteoblastic differentiation, was used to assess the anabolic response of MC3T3-E1 osteoblastic cells. When fluid forces were low, neither strain magnitude nor strain rate was correlated with OPN expression. Higher magnitudes of fluid shear stress, however, significantly increased OPN message levels independently of the strain magnitude or rate. The study suggests that fluid shear stress may play a more important role than strain in the bone response to mechanical loading.
Further evidence that strain alone is not the mechanotransducer comes from comparing in vivo to in vitro data. Customary strains in whole bone in vivo are typically in the range of 0.04% to 0.3% (400-3000 µϵ) for animal and human locomotion but seldom exceed 0.1% (1000 µϵ). Assuming that cell membrane stretch directly results from surrounding tissue deformation, strain on osteocyte/osteoblast membranes should be comparable to the bone tissue strain. However, in vitro studies show that to induce any cellular response by direct mechanical deformation of bone cells, deformations need to be one to two orders of magnitude larger than the bone tissue strains normally experienced by the whole bone in vivo. The larger strains needed to stimulate osteocytes/osteoblasts cannot be derived directly from matrix deformations because they would cause bone fracture.
A recent model by Weinbaum suggests that the amount of strain the osteoblastic cells experience in vivo may be amplified by the action of fluid flow on pericellular matrix and its coupling to the intracellular actin cytoskeleton. This model predicts that physiological levels of fluid shear stress could produce cellular levels of strain in bone up to 100-fold greater than normal levels of strain in tissues (0.04%-0.3%, or 400-3000 µϵ). Weinbaum concludes that the strain in the membrane of cell processes caused by the loading can be of the same order as the in vitro strains measured in cell culture studies where intracellular biochemical responses are observed for cells on stretched elastic substrates.
Mechanosensitive Cells
The PDL and bone contain a variety of cell types, and there is debate about which cells are mechanosensitive. Osteocytes, terminally differentiated osteoblasts housed in mineralized lacunae and communicating with each other via processes extending through narrow canaliculi, are considered to form the major strain-sensing network in bone. A theoretical model for flow-generated shear stresses in lacunar-canicular spaces developed by Weinbaum et al. predicts physiological fluid-induced shear stresses of 8 to 30 dynes/cm 2 in the proteoglycan-filled fluid annuli around osteocyte processes.
It is generally assumed that the marrow sinusoids enclosing osteoblasts are much too wide to generate meaningful levels of shear stress during physiological loading. However, recent studies have indicated that very low levels of shear stress are able to induce gene expression of a major enzyme needed for PGE 2 production, cyclooxygenase-2 (COX-2), in osteoblastic cells, suggesting a role for osteoblastic cells in the detection of mechanical forces. When rats are reloaded after 2 weeks of tail suspension, there is a transient increase on c-Fos expression in periosteal cells and an increase in COX-2 expression in osteocytic cells within the femur, suggesting that both osteoblasts and osteocytes are mechanosensitive.
Interestingly, bone marrow pre-osteoclasts and osteoclast cells may also be mechanosensitive, although the physiological significance of these effects remains unclear. Current thinking is that all or most cells are mechanosensitive, and their in vivo context determines the physiological significance of their responses to mechanical loading.
Mechanoreceptors
Bone and PDL cells must be able to convert external signals, fluid shear stress, and strain into intracellular signals. For this to occur, the PDL and bone cells must have a mechanism that is sensitive to external forces. Proposed mechanosensitive mechanisms include the integrin-cytoskeleton–nuclear matrix structure, G-protein–dependent pathways, stretch-activated ion channels within the cell membrane, and plasma disruption. Recent evidence suggests that the entire cell is a mechanosensor and that many different pathways are available for the transduction of a mechanical signal.
Integrins and tensegrity model
Integrins are the major family of cell surface receptors that mediate attachment to the extracellular matrix (ECM). They are composed of alpha (α)and beta (β)transmembrane subunits. There are currently 16 known α and eight known β subunits that heterodimerize to produce more than 20 different receptors. Most integrins bind ligands that are components of the extracellular matrices (e.g., collagen, fibronectin, vitronectin). These ligands cross-link or cluster integrins by binding to adjacent integrin molecules on the cell surface. The localized attachment domains within which integrin receptors cluster are referred to as focal adhesions. Focal adhesions form complexes that contain actin-associated proteins, such as talin, vinculin, paxillin and α-actinin. Focal adhesion complex proteins interact with the cytoplasmic portions of integrins and physically interconnect the ECM with the actin cytoskeleton. This structural interconnection not only serves as an anchor, but also is hypothesized to mediate mechanosensation ( Fig. 26-2 ). Also associated with these complexes are kinases, which can be either targets or initiators of various signaling pathways.
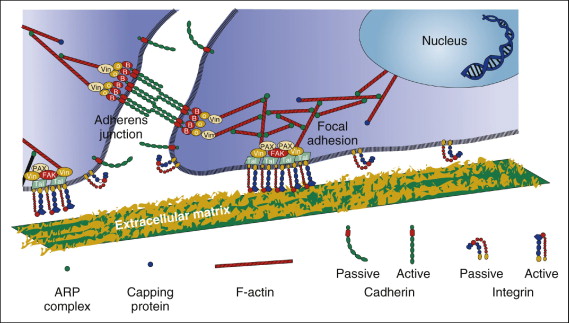
According to the tensegrity model, mechanical forces in the cell are balanced between tensile actin filaments, microtubular struts, and ECM anchoring supports. Integrins are responsible for maintaining the stability of this equilibrium. When external forces are applied to cells, the internal cellular tension changes to equalize the external forces by coordinate changes in actin bundle assembly. Prolonged exposure of cells to steady fluid flow results in their realignment in the direction of flow, a process driven by the rearrangement of the cytoskeleton. The actin cytoskeleton of cells exposed to flow changes from a disorganized banding pattern to almost-parallel fibers (stress fibers) aligned to the direction of flow. It is believed that actin cytoskeletal changes initiate protein phosphorylation cascades within focal contacts. Shear stress applied to the luminal surface of endothelial cells results in directional remodeling of abluminal focal adhesion sites and causes the activation of cellular signaling. Initiation of cell signaling involves a nonreceptor tyrosine kinase called focal adhesion kinase (FAK). FAK is tyrosine phosphorylated and localizes to focal adhesions after exposure to fluid shear stress. FAK activation by mechanical forces leads to activation of the mitogen-activated protein (MAP) kinase signaling pathway. Also, integrins are involved in fluid shear stress induction of new gene expression in osteoblastic cells.
Transmembrane G-protein receptor pathways
Another proposed mechanosensory pathway within the cell is through transmembrane G-protein receptors. G-protein receptors contain seven transmembrane hydrophobic domains associated with α, β, or gamma (γ) subunits, each encoded by separate gene families. The family that encodes the α subunit is especially diverse. In the resting state, guanine diphosphate (GDP) is bound to the α subunit. On binding of the ligand, the GDP is released and guanine triphosphate (GTP) is bound. This causes a conformational change and the disassociation of the α-GTP subunit from the β and γ subunits. The subunits then regulate metabolic pathways, resulting in the activation of various second messengers, enzymes, and ion channels. Inactivation is caused by hydrolysis of the GTP, which leads to reassociation of the α subunit with GDP and the β and γ subunits.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


