Chapter 25
Congenital Bleeding and Hypercoagulable Disorders
A number of procedures that are performed in dentistry may cause bleeding. Under normal circumstances, these procedures can be performed with little risk to the patient; however, the patient whose ability to control bleeding has been altered by congenital defects in coagulation factors, platelets, or blood vessels may be in grave danger unless the dentist identifies the problem before performing any dental procedure. In most cases, once the patient with a congenital bleeding problem has been identified, steps can be taken to greatly reduce the risks associated with dental procedures. The following disorders are discussed in this chapter: hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome), von Willebrand disease, Bernard-Soulier disease, Glanzmann’s thrombasthenia, hemophilia A, hemophilia B (Christmas disease), and congenital hypercoagulability disorders.
Definition
Inherited (congenital) bleeding disorders are genetically transmitted. They may involve a deficiency of one of the coagulation factors, abnormal construction of platelets, deficiency of von Willebrand factor, or malformation of vessels (< ?xml:namespace prefix = "mbp" />

Box 25-1 Classification of Congenital Bleeding and Thrombotic Disorders
Nonthrombocytopenic Purpuras
Disorders of Platelet Function
von Willebrand’s disease (may have secondary factor VIII deficiency)
Bernard-Soulier disease

Epidemiology: Incidence and Prevalence
The most common inherited bleeding disorder is von Willebrand disease. It affects about 1% of the U.S. population. The disease usually is inherited as an autosomal dominant trait. Hemophilia A, factor VIII deficiency, is the most common of the inherited coagulation bleeding disorders. It occurs in about 1 of every 5000 male births. More than 20,000 individuals in the United States have hemophilia A.
Etiology
Patients may be born with a deficiency of one of the factors needed for blood coagulation—for example, factor VIII deficiency as in hemophilia A or factor IX deficiency as in hemophilia B or Christmas disease. Congenital deficiencies of the other coagulation factors have been reported but are rare (
TABLE 25-1 Blood Coagulation Components
| Factor | Deficiency | Function |
|---|---|---|
| Factor II (prothrombin) | Congenital—rare | Protease zymogen |
| Factor X | Congenital—rare | Protease zymogen |
| Factor IX | Congenital—rare | Protease zymogen |
| Factor VII | Congenital—very rare | Protease zymogen |
| Factor VIII | Congenital—more common | Cofactor |
| Factor V | Congenital—rare | Cofactor |
| Factor XI | Congenital—rare | Protease zymogen |
| Factor XII | Deficiency reported but does not cause bleeding; aPTT will be prolonged |
Protease zymogen |
| Factor I (fibrinogen) | Congenital—rare | Structural |
| von Willebrand factor | Congenital—most common | Adhesion |
| Tissue factor | Not applicable | Cofactor initiator |
| Factor XIII | Congenital—rare; will cause bleeding, but aPTT and PT will be normal | Fibrin stabilization |
| High-molecular-weight kininogen | Deficiency does not cause bleeding; will prolong aPTT | Coenzyme |
| Prekallikrein | Deficiency does not cause bleeding; will prolong aPTT | Coenzyme |
Data from McVey JH: Coagulation factors. In Young NS, Gerson SL, High KA, editors: Clinical hematology, St. Louis, 2006, Mosby.
In von Willebrand disease, the primary problem involves lack of various sizes of von Willebrand factor (vWF), which are needed to attach platelets to damaged vascular wall tissues and to carry factor VIII in circulation.
HHT is a disorder consisting of multiple telangiectatic lesions involving the skin and mucous membranes.
Pathophysiology
The three phases of hemostasis for controlling bleeding are vascular, platelet, and coagulation. The vascular and platelet phases are referred to as primary, and the coagulation phase is secondary. The coagulation phase is followed by the fibrinolytic phase, during which the clot is dissolved. These hemostatic mechanisms are discussed in detail in
Clinical Presentation
Signs and Symptoms
The most common objective findings in patients with genetic coagulation disorders are ecchymoses, hemarthrosis, and dissecting hematomas (
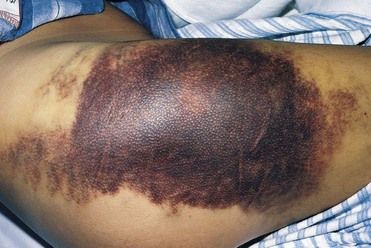
FIGURE 25-1 Large Area of Subcutaneous Ecchymoses Due to Trauma in a Patient with Hemophilia.
(From Hoffbrand AV, Pettit JE: Color atlas of clinical hematology, ed 4, London, 2010, Mosby.)
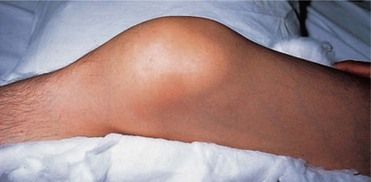
FIGURE 25-2 Acute Hemarthrosis of the Knee is a Common Complication of Hemophilia.
It may be confused with acute infection unless the patient’s coagulation disorder is known, because the knee is hot, red, swollen, and painful.
(From Forbes CD, Jackson WF: Color atlas and text of clinical medicine, ed 3, London, 2003, Mosby.)
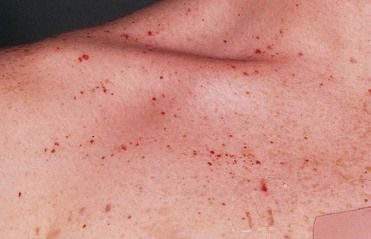
FIGURE 25-3 Purpura (Petechiae)- in this Case Throbocytopenia Purpura (TP).
The patient was a 15-year-old boy whose antiepileptic treatment regimen had recently been modified to include sodium valproate. This is just one of a number of drugs that may induce TP, but the disorder is almost always reversible if the drug therapy is stopped.
(From Forbes CD, Jackson WF: Color atlas and text of clinical medicine, ed 3, London, 2003, Mosby.)
Laboratory Tests
Three tests are recommended for use in initial screening for possible bleeding disorders
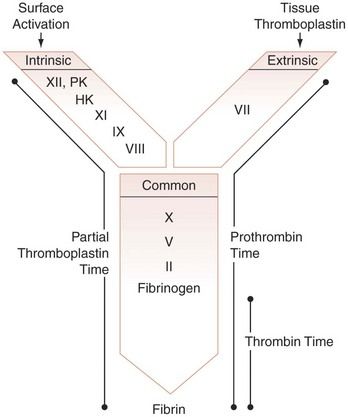
FIGURE 25-4 Organization of the coagulation system based on current screening assays. The intrinsic coagulation system consists of the protein factors XII, XI, IX, and VIII and prekallikrein (PK) and high-molecular-weight kininogen (HK). The extrinsic coagulation system consists of tissue factor and factor VII. The common pathway of the coagulation system consists of factors X, V, and II and fibrinogen (I). The activated partial thromboplastin time requires the presence of every protein except tissue factor and factor VII. The prothrombin time (PT) requires tissue factor; factors VII, X, V, and II; and fibrinogen. The thrombin clotting time only tests the integrity of fibrinogen.
(From McPherson RA, Pincus MR, editors: Henry’s clinical diagnosis and management by laboratory methods, ed 22, London, 2012, Saunders.)
Patients with positive results on screening tests should be evaluated further so that the specific deficiency can be identified and the presence of inhibitors ruled out. A hematologist orders these tests, establishes a diagnosis that is based on the additional testing, and makes recommendations for treatment of the patient who is found to have a significant bleeding problem. The screening laboratory tests are discussed in detail in
In patients with prolonged aPTT, PT, and TT, the defect involves the last stage of the common pathway, which is the activation of fibrinogen to form fibrin to stabilize the clot. The plasma level of fibrinogen is determined, and if it is within normal limits, then tests for fibrinolysis are performed. These tests, which detect the presence of fibrinogen and/or fibrin-degradation products, consist of staphylococcal clumping assay, agglutination of latex particles coated with antifibrinogen antibody, and euglobulin clot lysis time.
Medical Management
In this section, congenital conditions that may cause clinical bleeding are considered. The emphasis is placed on identification of patients with a potential bleeding problem and management of these patients if surgical procedures are needed.
TABLE 25-2 Medical Treatment of Congenital Bleeding Disorders
| Condition | Defect | Medical Management |
|---|---|---|
| Hereditary hemorrhagic telangiectasia | Multiple telangiectasias with mechanical fragility of the abnormal vessels | |
| von Willebrand disease | Deficiency or defect in vWF causing poor platelet adhesion and in some cases deficiency of F-VIII | |
| Hemophilia A | Deficiency or defect in F-VIII | |
| Some patients develop antibodies (inhibitors) to F-VIII | Porcine factor VIII, PCC, aPCC, factor VIIa, and/or steroids for patients with inhibitors | |
| Hemophilia B | Deficiency or defect in F-IX | |
| Development of antibodies (inhibitors) to F-IX is much less common than with hemophilia A | PCC, aPCC, factor VIIa, |
|
| Bernard-Soulier disease | Genetic defect in platelet membrane, absence of glycoprotein Ib (GP-Ib) causes disorder in platelet adhesion | |
| Glanzmann’s thrombasthenia | Genetic defect in platelet membrane, absence of glycoprotein IIb/IIIa (GPIIb/IIIa) |
aPCC, Activated prothrombin complex concentrates; PCC, Prothrombin complex concentrates; vWF, von Willebrand factor.
* Factor VIIa is activated factor VII.
TABLE 25-3 FDA-Approved Clotting Concentrates for Hemophilia A and B
| Preparation with Virucidal Technique(s) | Type/Manufacturer | Specific Activity (IU/mg Protein) |
|---|---|---|
| Ultrapure recombinant factor VIII | ||
| Immunoaffinity; ion exchange chromatography | Recombinate (Baxter) | >4000 |
| Ion exchange chromatography, nanofiltration | Refacto (Wyeth) | 11,200-15,000 |
| Ion exchange chromatography, ultrafiltration | Kogenate FS (Bayer Inc.) | >4000 |
| No human or animal protein used in culture; immunoaffinity and ion exchange chromatography | Advate (Baxter) | >4000-10,000 |
| Ultrapure human plasma factor VIII | ||
| Chromatography and pasteurization | Monoclate P (ZLB Behring) | >3000 |
| Chromatography and solvent detergent | Hemofil M (Baxter) | >3000 |
| High-purity human plasma factor VIII | ||
| Chromatography, solvent detergent, dry heating | Alphanate SD (Grifols) vWF | 50->400 |
| Solvent detergent, dry heating | Koate-DVI (Bayer) vWF | 50-100 |
| Pasteurization (heating in solution) | Humate-P (ZLB-Behring) vWF | 1-10 |
| Porcine plasma-derived factor VIII | ||
| Solvent detergent viral attenuation | Hyate-C (Ibsen/Biomeasure, Inc.) | >50 |
| Ultrapure recombinant factor IX | ||
| Affinity chromatography and ultrafiltration | BeneFix (Wyeth) | >200 |
| Very highly purified plasma factor IX | ||
| Chromatography and solvent detergent | AlphaNine SD (Grifols, Inc.) | >200 |
| Monoclonal antibody ultrafiltration | Mononine (ZLB-Behring, Inc.) | >160 |
| Low-purity plasma factor IX complex | ||
| Solvent detergent | Profilnine SD (Grifols, Inc.) | <50 |
| Vapor heat | Bebulin VH (Baxter) | <50 |
| Activated plasma factor IX complex concentrate (used primarily for patients with alloantibody and autoantibody factor VIII and IX inhibitor) | ||
| Vapor heat | FEIBA VH (Baxter) | <50 |
| Recombinate factor VIIa (indicated for patients with alloantibody and autoantibody factor VIII and IX inhibitors) | ||
| Affinity chromatography, solvent detergent | NovoSeven (Novo Nordisk, Inc.) | 50,000 |
FDA, U.S. Food and Drug Administration; vWF, Von Willebrand factor.
Data from Kessler CM: Hemorrhagic disorders: coagulation factor deficiencies. In Goldman L, Ausiello D, editors: Cecil medicine, ed 23, Philadelphia, 2008, Saunders.
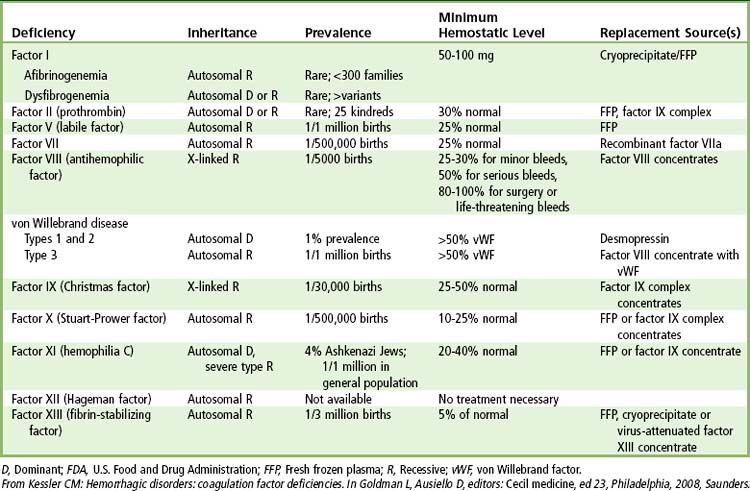
TABLE 25-4 FDA-Approved Coagulation Proteins and Replacement Therapies Available in the United States
Vascular Defects
HHT, also referred to as Osler-Weber-Rendu syndrome, is a rare autosomal dominant disorder that is characterized by multiple telangiectatic lesions involving the skin, mucous membranes, and viscera. One form of the disorder, characterized by a high frequency of symptomatic pulmonary arteriovenous malformations and cerebral abscesses, has been identified as being due to abnormalities in the endothelial protein endoglin (ENG), encoded by the gene HHT1, located on chromosome 9. A second form of the disease has been linked to the activin receptor-like kinase 1 gene located on chromosome 12 (HHT2). Other forms of the disease have been linked to loci on chromosome 5q (HHT3), 7p14 (HHT4), and a region of chromosome 3 encoding the TGF-βII receptor (HHT5).
The telangiectasias consist of focal dilation of postcapillary venules with connections to dilated arterioles, initially through capillaries and later directly. Perivascular mononuclear cell infiltrates also are observed. The vessels of HHT show a discontinuous endothelium and an incomplete smooth muscle cell layer. The surrounding stroma lacks elastin. Thus, the bleeding tendencies are thought to be due to mechanical fragility of the abnormal vessels.
Clinical Findings
On clinical examination, venous lakes and papular, punctate, matlike, and linear telangiectasias appear on all areas of the skin and mucous membranes, with a predominance of lesions on and under the tongue and on the face, lips, perioral region, nasal mucosa, fingertips, toes, and trunk.
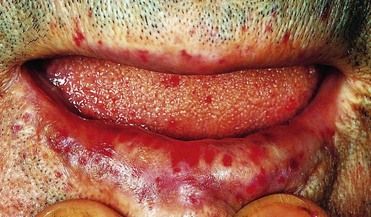
FIGURE 25-5 Hereditary Haemorrhagic Telangiectasia (HHT).
HHT is a condition in which occult blood loss in the gut may lead to severe iron deficiency anemia. The diagnosis usually is clear from a careful clinical examination, although the telangiectases are not always as obvious, as in this patient with multiple lesions on the face, lip, and tongue. The patient had received multiple blood transfusions over many years because of HHT-associated gastrointestinal blood loss, and he had developed cirrhosis associated with hepatitis B antigen positivity—probably as a result of transmission of hepatitis B in transfused blood.
(From Forbes CD, Jackson WF: Color atlas and text of clinical medicine, ed 3, London, 2003, Mosby.)
Laboratory Tests
There are no reliable laboratory tests to determine the tendency for bleeding to occur in affected persons. The Ivy bleeding time is not useful. Clinical findings and a history of bleeding problems are the only effective means to identify patients at risk.
Treatment
Therapy for HHT remains fragmented and problematic, consisting of laser treatment for cutaneous lesions; split-thickness skin grafting, embolization of arteriovenous communications, or hormonal therapy (estrogen or estrogens plus progesterone) for epistaxis; pulmonary resection or embolization for pulmonary arteriovenous malformations; and hormonal therapy and laser coagulation for gastrointestinal lesions.
The nasal vasculature pattern may help to predict the response to laser therapy versus septodermaplasty. Resurfacing the nasomaxillary cavity with radial forearm fasciocutaneous free flaps has been reported to be effective in patients with refractory epistaxis. The antifibrinolytic agents aminocaproic acid and tranexamic acid have been reported to be beneficial in controlling hemorrhage, but negative results with antifibrinolytic therapy also have been reported. Improvement in lesions has been reported in cases using an antagonist to vascular endothelial growth factor and sirolimus and aspirin. Patients with gastrointestinal bleeding should receive supplemental iron and folate; red blood cell transfusions and parenteral iron may be required in some patients.
Platelet Disorders
von Willebrand Disease
The most common inherited bleeding disorder is von Willebrand disease, which is caused by an inherited defect involving platelet adhesion. The vWF gene and protein and points of mutation are shown in
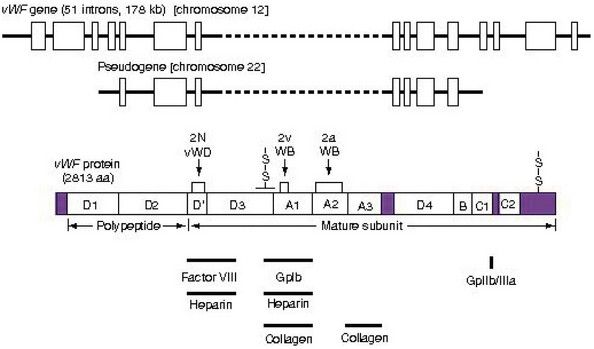
FIGURE 25-6 The structure of the vWF gene and protein. The structures of the vWF gene and pseudogene are indicated schematically at the top of the figure. The corresponding protein also is depicted, including the homologous repeat domain structure. The localization within vWF of point mutations associated with VWD variants also is indicated. AA, Amino acids; vWD, von Willebrand disease; vWF, von Willebrand factor.
(From Hoffman R, et al, editors: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone.)
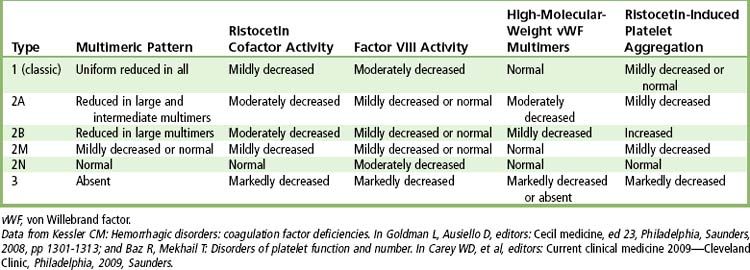
The disease has several variants, depending on the severity of genetic expression (
Clinical Findings
Mild variants of von Willebrand disease are characterized by a history of cutaneous and mucosal bleeding because platelet adhesion is lacking. In the more severe forms of the disease, in which factor VIII levels are low, hemarthroses and dissecting intramuscular hematomas are part of the clinical picture. Petechiae are rare in these patients. However, gastrointestinal bleeding, epistaxis, and menorrhagia are very common.
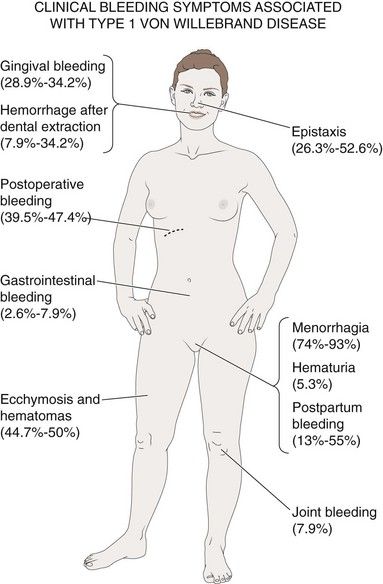
Laboratory Tests
Laboratory investigation is needed to make the diagnosis. Screening laboratory tests may show prolonged aPTT, normal or slightly reduced platelet count, normal PT, and normal TT. Additional laboratory tests are needed to establish the diagnosis and type of von Willebrand disease. These consist of ristocetin cofactor activity, ristocetin-induced platelet aggregation, immunoassay of vWF, multimeric analysis of vWF, and specific assays for factor VIII.
Treatment
Treatment depends on the clinical condition of the patient and the type of von Willebrand disease that is diagnosed. Available treatment options include cryoprecipitate, factor VIII concentrates that retain HMW vWF multimers (Humate-P, Koate HS), and desmopressin (1-deamino-8-d-arginine vasopressin [DDAVP]). Before desmopressin is given, the patient must be tested for response to the agent as some patients are nonresponsive. Desmopressin can be given parenterally or by nasal spray 1 hour before surgery. With parenteral administration, the dose of desmopressin is 0.3 µg/kg of body weight, with a maximum dose of 20 to 24 µg. The nasal spray, Stimate, contains 1.5 mg/mL of desmopressin and is given at a dose of 300 mg/kg. Usually, one dose is sufficient. If a second dose is needed, it is given 8 to 24 hours after the first dose. Desmopressin should be used with caution in older patients with cardiovascular disease because of the potential risk of drug-induced thrombosis.
Patients with type 1 von Willebrand disease are the best candidates for desmopressin therapy. Desmopressin treatment must not be started without previous testing to determine which variant form of von Willebrand disease is involved. It is not effective for type 3 and most variants of type 2 von Willebrand disease. These patients are treated with factor VIII replacement that retains the HMW (high molecular weight) vWF multimers (Humate-P or Koate HS). In patients with type 2 variants with qualitative defects in vWF, Humate-P or Koate HS supplies functional HMW vWF and factor VIII for those with/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


