Treacher Collins syndrome (TCS), or mandibulofacial dysostosis, is an autosomal dominant condition with variable expressivity ( Figure 47-1 ). It generally is characterized by bilaterally symmetric abnormalities of structures within the first and second branchial arches. Early descriptions are attributed to Berry (1889), Treacher Collins (1900), and Franceschetti and Klein (1949). To date, the physical findings of many hundreds of individuals and families with TCS have been published in the world literature.
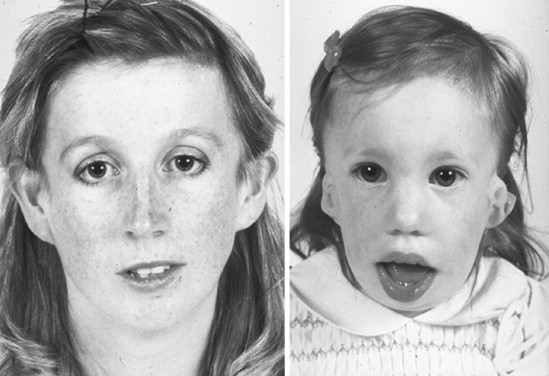
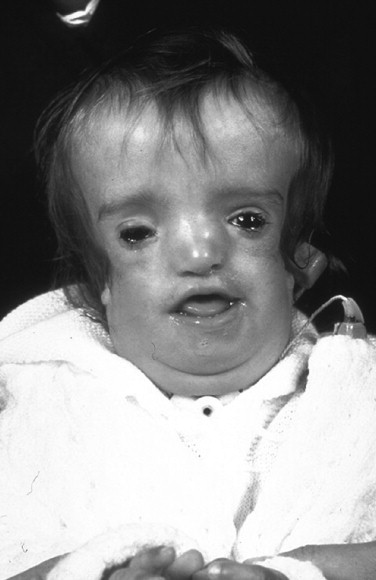
The adult patient with fully expressed TCS has a convex facial profile with a prominent nasal dorsum above a retrusive lower jaw and chin. The eyes are characterized by an anti-mongoloid slant of the palpebral fissure, resulting from colobomata and hypoplasia of the lower lids and lateral canthi, including partial absence of eyelid cilia and inferolateral orbital hypoplasia/dystopia ( Figure 47-2 ). “Tongue-shaped” processes of hair frequently extend into the preauricular region. The external ears are absent, malformed, or malposed, and hearing is impaired as a result of variable degrees of hypoplasia of the external auditory canals and ossicles of the middle ears. A characteristic finding is hypoplasia of the malar bones, often with clefting through the arches and limited formation of the residual zygomas, including the glenoid fossa. The maxilla and mandible bones are also characteristically hypoplastic, with variable effects on the temporomandibular joints (TMJs), the muscles of mastication, and the muscles of facial expression. It is interesting to note that hypoplasia of the centrally located soft tissues of the face is not seen in TCS. In general, an Angle class II anterior open-bite malocclusion and a steep (rotated clockwise) occlusal plane are present. The presence of cleft palate, with or without cleft lip and choanal atresia, is variable. Dental anomalies, including tooth agenesis (33.3%), enamel opacities (20%), and ectopic eruption of the maxillary first molars (13.3%), are present in 60% of individuals.
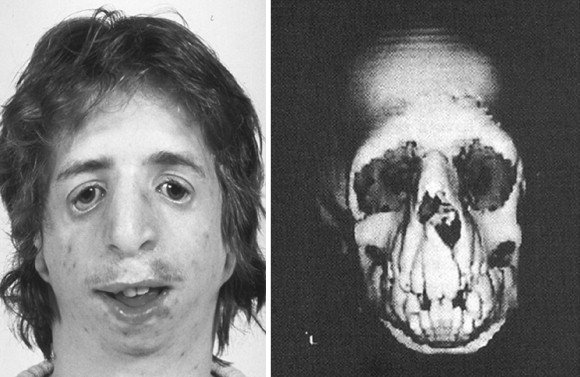
Patients with TCS have a reduced cranial base angle (basilar kyphosis). Craniosynostosis is not a feature of TCS, but the neurocranium may have an abnormal shape (decreased anteroposterior length and diminished bitemporal width) that is evident during childhood and remains so through adulthood. The degree of malformation present at birth is believed to be relatively stable and nonprogressive with age.
INHERITANCE, GENETIC MARKERS, AND TESTING
Treacher Collins syndrome occurs in 1 in 25,000 to 1 in 50,000 live births. Inheritance is autosomal dominant, and males and females are equally affected. About 60% of probands with TCS have the disorder as the result of a de novo gene mutation. Each child of an individual with TCS has a 50% chance of inheriting the mutation. TCOF1 is the only gene currently known to be associated with TCS. Direct sequencing of the coding and flanking intronic regions of TCOF1 defects and mutations occurs in about 90% to 95% of individuals.
Genetic counseling for TCS is the process of providing individuals and families with information on the nature, inheritance, and implications of the syndrome to help them make informed medical and personal decisions. Molecular testing should be considered for any individual who presents with at least two major features or three minor features of TCS. Until recently, penetrance in TCS was believed to be complete. Marres et al (1995) confirmed the absence of clinical and radiographic findings in an individual with a pathogenic TCS TCOF1 mutation. For this reason, testing is acceptable for individuals with any degree of severity of TCS features, to confirm or refute the diagnosis.
Prenatal diagnosis of pregnancies at increased risk for TCS is possible through analysis of DNA extracted from fetal cells obtained by amniocentesis performed at about 15 to 18 weeks’ gestation or by chorionic villus sampling (CVS) at about 10 to 12 weeks’ gestation. The malformation-causing allele of an affected family member must be identified before prenatal testing can be performed.
DYSMORPHOLOGY
CRANIO-ORBITOZYGOMATIC REGION
In earlier published studies, 14 reproducible cranio-orbitozygomatic measurements were taken from each of 26 standard axial computed tomographic (CT) scans of individuals with symmetric forms of TCS who had not been operated on, and values were compared with age-matched controls. The interorbital measurements (medial and lateral orbital wall separations) of patients with TCS were at the mean when compared with the cohort group, whereas the zygomatic measurements were significantly lower than normal, confirming the extent of malar hypoplasia. The congenitally deficient lateral aspect of the orbits in individuals with TCS was confirmed by the greater than normal values measured for globe protrusion and medial orbital wall protrusion in conjunction with diminished lateral orbital wall lengths, all of which use the lateral orbital rim as a reference point. The abnormal shape of the anterior cranial vault in patients with TCS was documented as a diminished intercoronal (bitemporal) distance (width) and decreased cephalic length when compared with values for normal, age-matched controls.
In general, the CT measurements documented in our study are consistent with the clinically observed morphology of TCS. Measurements carried out in the zygomatic structures confirmed the extent of hypoplasia of the zygomas for the group as a whole and in each individual patient. They also documented the extent of global protrusion and decreased upper face width in patients with TCS.
MAXILLOMANDIBULAR REGION
Roberts and co-workers, in 1975, completed a radiographic cephalometric study of TCS. Serial cephalometric radiographs were obtained in eight patients who presented with the full range of features characteristic of this syndrome. The expected facial convexity in TCS was substantiated by cephalometric measurements. The extent of convexity was attributed to the severity of mandibular retrognathia (decreased sella–nasion–B point [SNB] angle), and the relationship of the maxilla to the cranial base (SNA angle) remained within normal limits among patients who were measured. On a longitudinal basis, the facial convexity remained relatively constant, confirming that the facial profile of an infant with TCS is remarkably similar to that of an adult. Anterior facial heights were measured as “upper facial height” (nasion–anterior nasal spine [N-ANS]) and “total facial height” (nasion–menton [N-M]). The upper facial height was found to be relatively normal, whereas the total facial height was often excessive; this was thought to be the result of a combination of anterior open-bite malocclusion, mandibular retrognathism, and chin dysplasia characterized by increased vertical length and horizontal retrusion.
The angle between the sella–nasion (S-N) plane and the palatal plane was obtuse, confirming the steepness (clockwise rotation) of the occlusal plane. Posterior facial height was markedly shorter for patients with TCS, with values in five of the eight patients found to be a minimum of 4 (standard deviation, SD) below those in the normal cohort group.
The effective horizontal length of the mandible was markedly reduced, with values in six of the eight patients falling below 4 SD when compared with those in the normal cohort group. Decreased mandible size was evident in both ramus height and body length. The gonial angle was confirmed to be obtuse, with values of 2 SD from the mean when compared with control values. The extent of “antigonial notching” was documented in patients with TCS and was found to be similar to that observed in patients with condylar arrest early in life (e.g., as a result of juvenile rheumatoid arthritis or TMJ trauma at a young age).
SOFT TISSUES
Soft tissue deficiencies and/or deformities in the patient with TCS are manifested primarily in four clinical regions: external ears, eyelid-adnexal structures, preauricular-cheek skin, and temporal fossa (e.g., temporalis muscle hypoplasia). The extent of soft tissue deficiency within the facial soft tissue envelope is variable and, to a certain extent, reflective of skeletal involvement. Each patient is unique and must be evaluated individually. Soft tissue deficiencies continue to represent the most difficult obstacle to favorable reconstruction.
EXTERNAL AUDITORY CANAL, MIDDLE EAR, AND AUDIOLOGIC FINDINGS
Relatively symmetric outer and middle ear malformations are characteristic of TCS. Middle ear abnormalities (e.g., hypoplastic or absent cavities, ossicles) are an almost universal finding. Most patients have a unilateral or bilateral moderate or greater conductive hearing loss, with a flat or reverse sloping configuration on the audiogram. Most of the hearing loss in these patients is attributable to middle ear malformations. Pron and colleagues, in 1993, reported the results of a detailed CT radiologic and audiologic investigation of a large series of pediatric patients with TCS. Objectives of the study were (1) to determine the degree and symmetry of the external auditory canal and middle ear abnormalities, and to evaluate hearing loss, and (2) to explore the relationship between hearing loss and external auditory canal and middle ear abnormalities in patients with TCS. Data were available from 29 children who had not yet undergone any ear interventions and who had undergone audiometric testing and focused standardized petrous (temporal) bone CT scans. A review of the CT scans permitted evaluation of the external ear canal and middle ear anatomy. Most patients (88%) had largely symmetric external auditory canal abnormalities that were stenotic (31%), atretic (54%), or normal (15%). Middle ear cavity ossicular deformities were generally symmetrical (96%), and cavities were hypoplastic (85%) or missing (4%). Cavities that were hypoplastic were also deformed, assuming a rectangular rather than an oval shape. Ossicle deformities were generally completely symmetric and consisted largely of hypoplastic (46%) or missing (46%) ossicles. Ossicles, both of the malleus and the incus, that were hypoplastic also tended to be ankylosed (82%) to the lateral or medial wall of the tympanic recess. Structures of the external auditory canal and middle ear generally are related. Increasing degrees of external ear canal malformation (normal/stenotic/atretic) are directly associated with ossicle and cavity malformations. Of the atretic canals, 67% had missing ossicles and 33% had small or ankylosed ossicles. No abnormalities were noted in the inner ear structures (i.e., cochlea, vestibule, canals, and internal auditory meatus) in patients with TCS.
According to the study of Pron and co-workers, hearing loss for patients with TCS ranges from mild to severe (average, 58 dB hearing loss [HL]). Most patients (96%) have a moderate or greater degree of hearing loss.
Relationships exist between external auditory canal and middle ear morphology and the degree and configuration of hearing loss. Although an overall direct association has been noted between increasing external ear canal malformation and degree of hearing loss in patients with TCS, hypoplastic, ankylosed, and missing ossicles appear to be associated with increasing, although not clinically significant, levels of hearing loss (on average, 52, 56, and 60 dB HL, respectively).
DYSMORPHOLOGY
CRANIO-ORBITOZYGOMATIC REGION
In earlier published studies, 14 reproducible cranio-orbitozygomatic measurements were taken from each of 26 standard axial computed tomographic (CT) scans of individuals with symmetric forms of TCS who had not been operated on, and values were compared with age-matched controls. The interorbital measurements (medial and lateral orbital wall separations) of patients with TCS were at the mean when compared with the cohort group, whereas the zygomatic measurements were significantly lower than normal, confirming the extent of malar hypoplasia. The congenitally deficient lateral aspect of the orbits in individuals with TCS was confirmed by the greater than normal values measured for globe protrusion and medial orbital wall protrusion in conjunction with diminished lateral orbital wall lengths, all of which use the lateral orbital rim as a reference point. The abnormal shape of the anterior cranial vault in patients with TCS was documented as a diminished intercoronal (bitemporal) distance (width) and decreased cephalic length when compared with values for normal, age-matched controls.
In general, the CT measurements documented in our study are consistent with the clinically observed morphology of TCS. Measurements carried out in the zygomatic structures confirmed the extent of hypoplasia of the zygomas for the group as a whole and in each individual patient. They also documented the extent of global protrusion and decreased upper face width in patients with TCS.
MAXILLOMANDIBULAR REGION
Roberts and co-workers, in 1975, completed a radiographic cephalometric study of TCS. Serial cephalometric radiographs were obtained in eight patients who presented with the full range of features characteristic of this syndrome. The expected facial convexity in TCS was substantiated by cephalometric measurements. The extent of convexity was attributed to the severity of mandibular retrognathia (decreased sella–nasion–B point [SNB] angle), and the relationship of the maxilla to the cranial base (SNA angle) remained within normal limits among patients who were measured. On a longitudinal basis, the facial convexity remained relatively constant, confirming that the facial profile of an infant with TCS is remarkably similar to that of an adult. Anterior facial heights were measured as “upper facial height” (nasion–anterior nasal spine [N-ANS]) and “total facial height” (nasion–menton [N-M]). The upper facial height was found to be relatively normal, whereas the total facial height was often excessive; this was thought to be the result of a combination of anterior open-bite malocclusion, mandibular retrognathism, and chin dysplasia characterized by increased vertical length and horizontal retrusion.
The angle between the sella–nasion (S-N) plane and the palatal plane was obtuse, confirming the steepness (clockwise rotation) of the occlusal plane. Posterior facial height was markedly shorter for patients with TCS, with values in five of the eight patients found to be a minimum of 4 (standard deviation, SD) below those in the normal cohort group.
The effective horizontal length of the mandible was markedly reduced, with values in six of the eight patients falling below 4 SD when compared with those in the normal cohort group. Decreased mandible size was evident in both ramus height and body length. The gonial angle was confirmed to be obtuse, with values of 2 SD from the mean when compared with control values. The extent of “antigonial notching” was documented in patients with TCS and was found to be similar to that observed in patients with condylar arrest early in life (e.g., as a result of juvenile rheumatoid arthritis or TMJ trauma at a young age).
SOFT TISSUES
Soft tissue deficiencies and/or deformities in the patient with TCS are manifested primarily in four clinical regions: external ears, eyelid-adnexal structures, preauricular-cheek skin, and temporal fossa (e.g., temporalis muscle hypoplasia). The extent of soft tissue deficiency within the facial soft tissue envelope is variable and, to a certain extent, reflective of skeletal involvement. Each patient is unique and must be evaluated individually. Soft tissue deficiencies continue to represent the most difficult obstacle to favorable reconstruction.
EXTERNAL AUDITORY CANAL, MIDDLE EAR, AND AUDIOLOGIC FINDINGS
Relatively symmetric outer and middle ear malformations are characteristic of TCS. Middle ear abnormalities (e.g., hypoplastic or absent cavities, ossicles) are an almost universal finding. Most patients have a unilateral or bilateral moderate or greater conductive hearing loss, with a flat or reverse sloping configuration on the audiogram. Most of the hearing loss in these patients is attributable to middle ear malformations. Pron and colleagues, in 1993, reported the results of a detailed CT radiologic and audiologic investigation of a large series of pediatric patients with TCS. Objectives of the study were (1) to determine the degree and symmetry of the external auditory canal and middle ear abnormalities, and to evaluate hearing loss, and (2) to explore the relationship between hearing loss and external auditory canal and middle ear abnormalities in patients with TCS. Data were available from 29 children who had not yet undergone any ear interventions and who had undergone audiometric testing and focused standardized petrous (temporal) bone CT scans. A review of the CT scans permitted evaluation of the external ear canal and middle ear anatomy. Most patients (88%) had largely symmetric external auditory canal abnormalities that were stenotic (31%), atretic (54%), or normal (15%). Middle ear cavity ossicular deformities were generally symmetrical (96%), and cavities were hypoplastic (85%) or missing (4%). Cavities that were hypoplastic were also deformed, assuming a rectangular rather than an oval shape. Ossicle deformities were generally completely symmetric and consisted largely of hypoplastic (46%) or missing (46%) ossicles. Ossicles, both of the malleus and the incus, that were hypoplastic also tended to be ankylosed (82%) to the lateral or medial wall of the tympanic recess. Structures of the external auditory canal and middle ear generally are related. Increasing degrees of external ear canal malformation (normal/stenotic/atretic) are directly associated with ossicle and cavity malformations. Of the atretic canals, 67% had missing ossicles and 33% had small or ankylosed ossicles. No abnormalities were noted in the inner ear structures (i.e., cochlea, vestibule, canals, and internal auditory meatus) in patients with TCS.
According to the study of Pron and co-workers, hearing loss for patients with TCS ranges from mild to severe (average, 58 dB hearing loss [HL]). Most patients (96%) have a moderate or greater degree of hearing loss.
Relationships exist between external auditory canal and middle ear morphology and the degree and configuration of hearing loss. Although an overall direct association has been noted between increasing external ear canal malformation and degree of hearing loss in patients with TCS, hypoplastic, ankylosed, and missing ossicles appear to be associated with increasing, although not clinically significant, levels of hearing loss (on average, 52, 56, and 60 dB HL, respectively).
FACIAL GROWTH POTENTIAL
The degree of malformation present at the time of birth in a newborn with TCS is believed to be relatively stable and nonprogressive with age. Roberts and associates reviewed serial cephalometric radiographs of patients with TCS and documented the stability of the inferior border of the mandible over time. Garner reported his findings from the cephalometric analysis of three patients of varied ages confirmed to have TCS. He documented the relative stability of the deformities observed at varied ages.
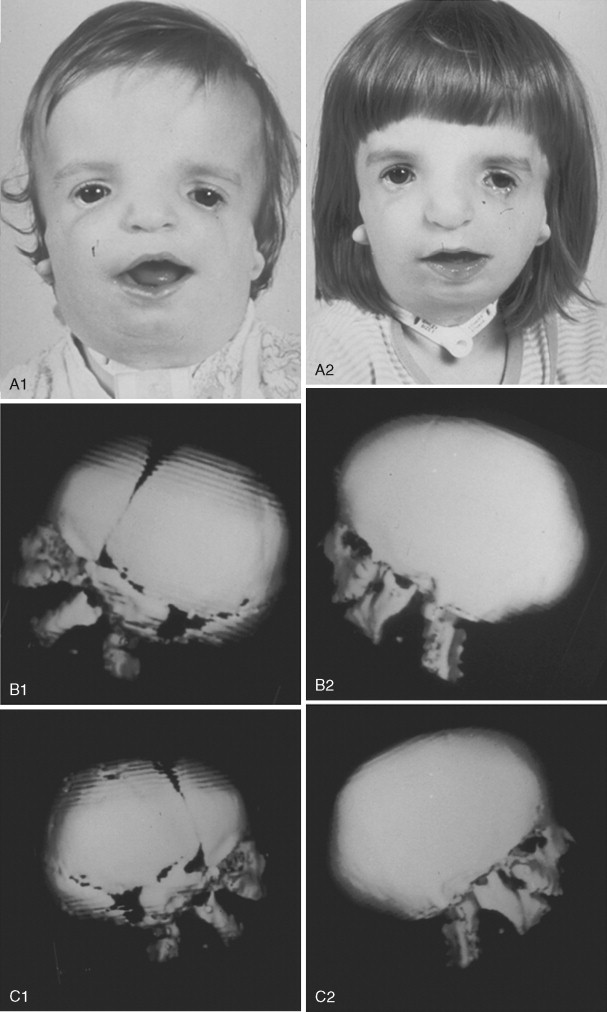
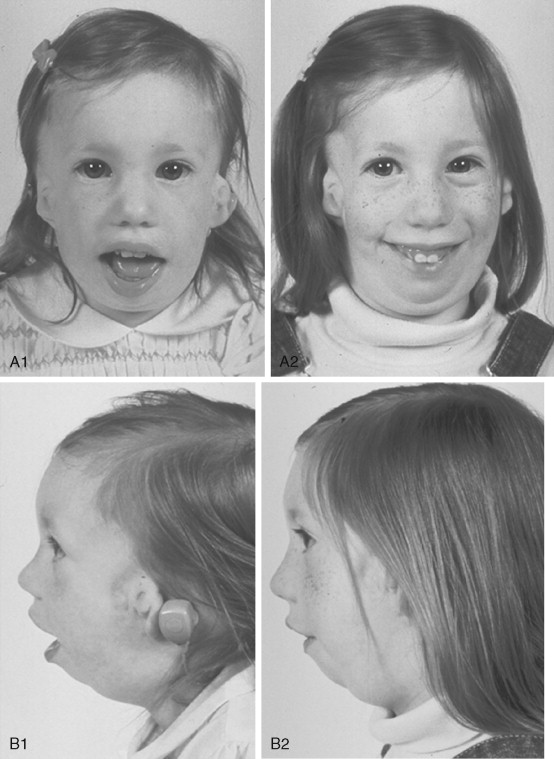
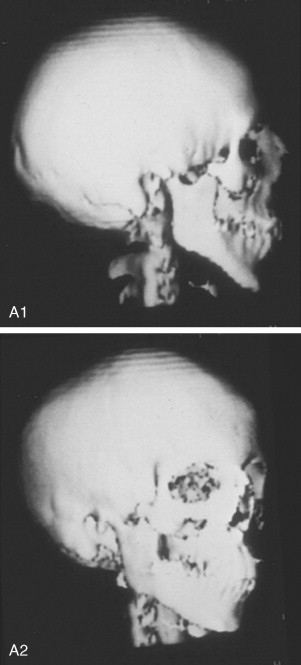
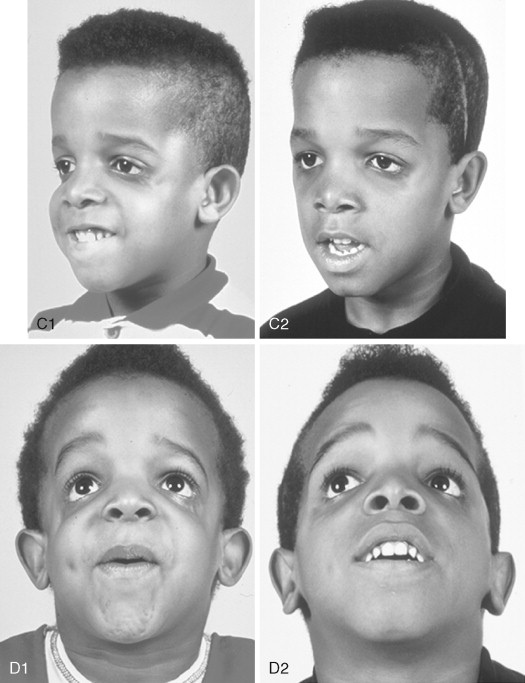
To date, no convincing evidence has been presented to demonstrate a clinically significant worsening of TCS dysmorphology with facial growth ( Figures 47-3 and 47-4 ). Early interventions with jaw osteotomies in which distraction osteogenesis or classic orthognathic techniques were used may adversely affect growth potential, necessitating additional revision osteotomies later. For this reason, if feasible, it is advisable to consider waiting until most growth is complete, prior to reconstruction of the skeletal components. In any case, “early” mandibular surgery would not be expected to limit the need for final maxillofacial reconstruction.
CONSIDERATIONS DURING INFANCY AND EARLY CHILDHOOD
At the time of birth of a child with TCS, concerns center on adequacy of the airway, swallowing, feeding, hearing, vision, presence of cleft palate (with or without cleft lip), and any associated malformations. The airway may be compromised as a result of two factors. The first is maxillary hypoplasia, with a degree of choanal stenosis or atresia, which constricts the nasal passages. In addition, the presence of mandibular micrognathia with a retropositioned tongue will at least partially obstruct the oropharyngeal and hypopharyngeal spaces. Depending on the severity of the deformities, a spectrum of airway difficulties may be present, necessitating special infant positioning, extended hospital stay, and pulse oximetry monitoring. Surgical considerations for management of airway compromise may include tongue–lip adhesion, immediate or delayed tracheostomy, or a mandibular advancement procedure.
As a result of the decreased efficiency of nasal and oral breathing and musculoskeletal malformations, the newborn may have difficulty with swallowing and achieving an adequate lip seal, to the extent that gavage-assisted feedings or placement of a gastrostomy tube is required to ensure adequate nutrition.
When a cleft of the secondary palate is documented, special attention must be given to the timing of palate closure and its additive effects on the airway. When Nager syndrome is present, the soft palate not only is clefted, it also is severely hypoplastic, to the extent that soft tissue is inadequate to achieve a functioning palate ( Figure 47-5 ). A primary pharyngeal flap, although helpful for velopharyngeal function, is rarely indicated because marked obstruction of the airway would likely result.
Within a few days of birth, results of examination by an experienced pediatric otolaryngologist in combination with formal audiology testing findings will clarify the extent of conductive hearing loss and allow for the early fitting of hearing aids to assist the infant with the acquisition of communication skills and encourage the normal bonding process with the family.
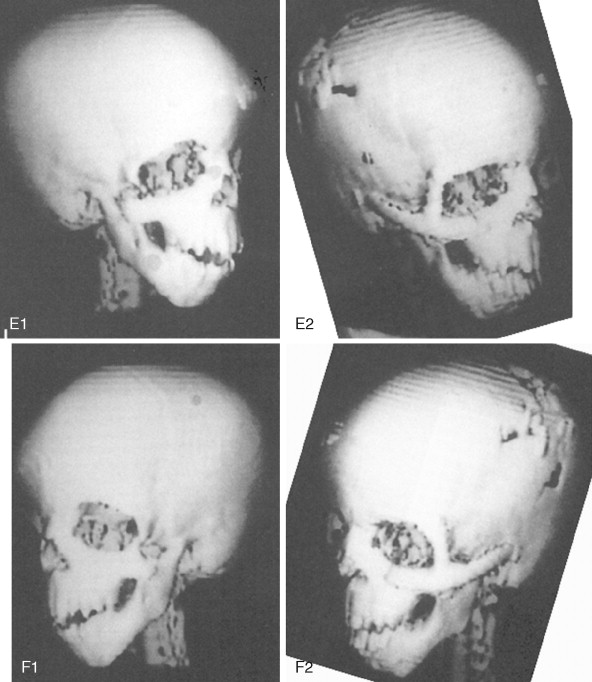
At some point early in life, a complete craniofacial CT scan (axial and coronal slices) from the top of the skull through the cervical spine, with three-dimensional re-formation, is useful for documenting the extent of craniofacial skeletal dysmorphology (see Figure 47-3 ). Special (focused) cuts through the petrous (temporal) bones are required to document external auditory canal and middle and inner ear anatomy.
A pediatric ophthalmologic assessment is necessary to determine extraocular muscle function, corneal exposure difficulties, and visual acuity. Ophthalmologic defects may include vision loss (37%), amblyopia (33%), refractive errors (58%), anisometropia (17%), and strabismus (37%). Evaluation by a medical geneticist (dysmorphologist) is indicated to document other possible malformations and to guide family planning. Investigators also found that family-to-family support is of great value.
It has been the subjective impression of many clinicians and scientists that the individual with TCS has a tendency toward short stature, at least early in life, and that above average intelligence is generally observed.
CLASSIFICATION OF TEMPOROMANDIBULAR JOINT–MANDIBULAR MALFORMATION
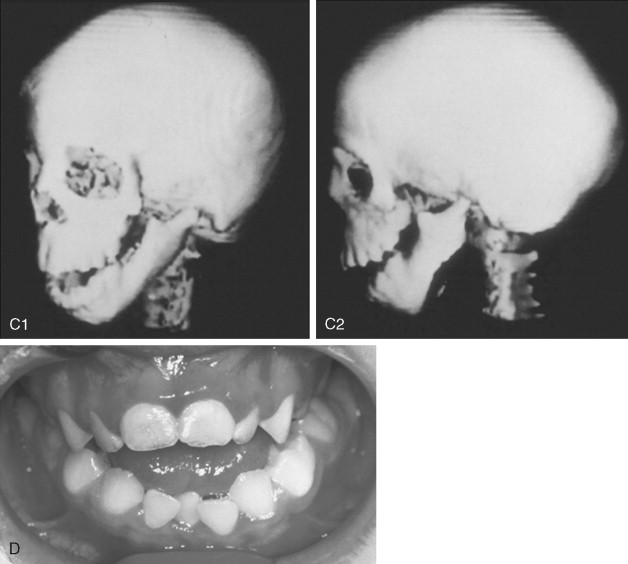
The extent of TMJ–mandibular malformation greatly influences the timing and techniques of mandibular reconstruction. Imprecise definitions for each type of mandibular malformation have prevented the consistent categorization of glenoid fossa–TMJ–ascending ramus deficiencies and have resulted in confusion and miscommunication. Kaban and co-workers designed a classification system for the degree of TMJ–mandibular deficiency observed in hemifacial microsomia. This classification is also useful for defining mandibular dysmorphology in the patient with TCS ( Figure 47-6 ). Even when the Kaban classification is used, it is important for the clinician to clearly define each type to ensure effective communication.
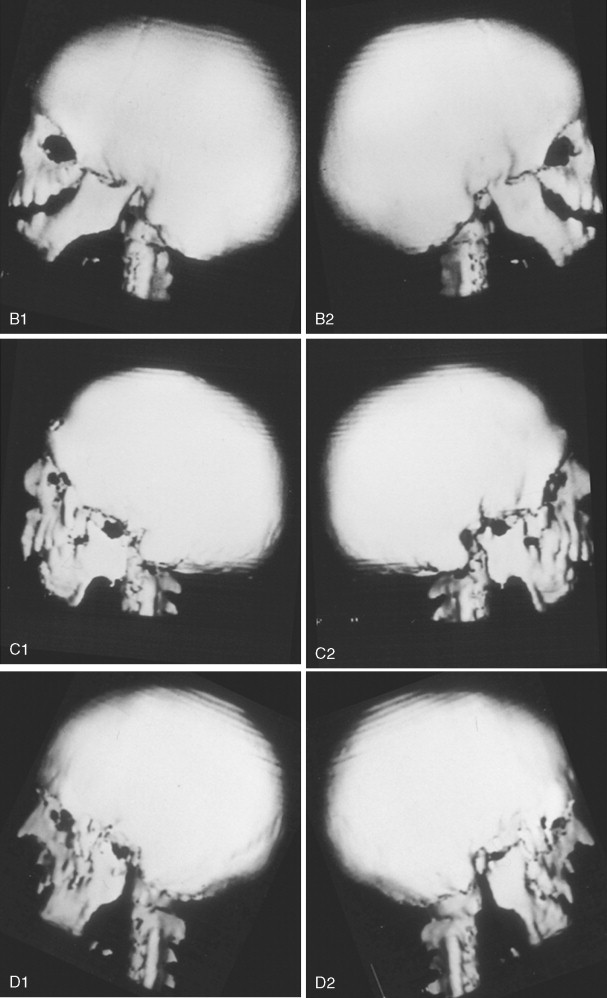
A type I mandible exhibits minimal hypoplasia of the glenoid fossa and the condyle–ascending ramus. Retrognathia and a mild anterior open bite may be present, but TMJ function is essentially normal ( Figure 47-6, A ). The muscles of mastication are intact.
A type IIA mandible demonstrates a moderate degree of glenoid fossa and condyle–ascending ramus hypoplasia, but TMJ function remains good ( Figure 47-6, B ). The location of the TMJ is somewhat anterior and medial. The condyle and the glenoid fossa are deemed acceptable and functioning at a level better than could be achieved through TMJ replacement. One or more of the paired muscles of mastication are hypoplastic, but all are present.
The type IIB mandible has moderate to severe hypoplasia of the condyle–ascending ramus and frequently the glenoid fossa ( Figure 47-6, C ). The TMJ–ascending ramus complex is anteriorly and medially located. The mandible does not have a consistent centric relation (CR), and the hypoplastic condyle is not consistently seated in the glenoid fossa. The mandible functions within a limited range of motion, and the extent of mandibular retrognathia and anterior open bite is marked. When posterior and superior pressure is applied to the chin if the condyles can be seated in a consistent location against/within each glenoid fossa/skull base, then ramus osteotomies will allow the proximal segments to be positioned into a stable location within the glenoid fossa/skull base location. Coronoidectomies may be required to adjust for the short condyles. In other patients with a type IIB mandibular deformity, the condyle is too far medially and anteriorly displaced or rudimentary to be useful, and construction of the condyle–ascending ramus with the use of a bone graft is required. Fortunately, in TCS, this is rarely required.
The type III mandible is free floating, with no “posterior stop” of the lower jaw against the cranial base ( Figure 47-6, D ). The condyle and the ascending ramus are not present; the disc, TMJ capsule, and glenoid fossa also are not developed. Severe mandibular dysmorphology, including (horizontal) retrognathia and decreased posterior facial (ramus) height, is present. A tracheostomy generally is required shortly after birth, to manage the airway. Eventual construction of both a glenoid fossa and a condyle–ascending ramus on each side with bone grafts will be required.
STAGING OF CRANIOFACIAL RECONSTRUCTION: TIMING AND TECHNIQUES
Craniofacial rehabilitation of the Treacher Collins malformation must address unique and specific components of the deformity, including zygomatic and orbital regions, maxillomandibular region, nasal region, soft tissues, external ears, and auditory canals and middle ear structures.
ZYGOMATIC AND ORBITAL RECONSTRUCTION
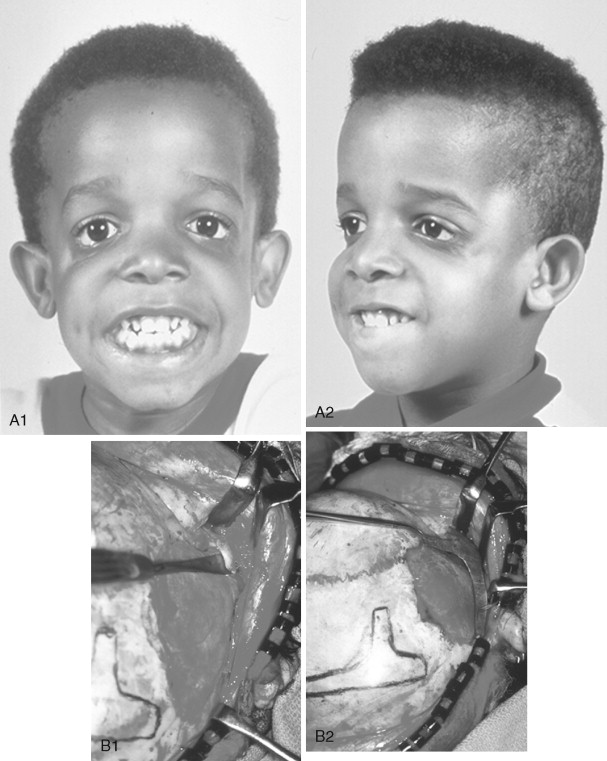
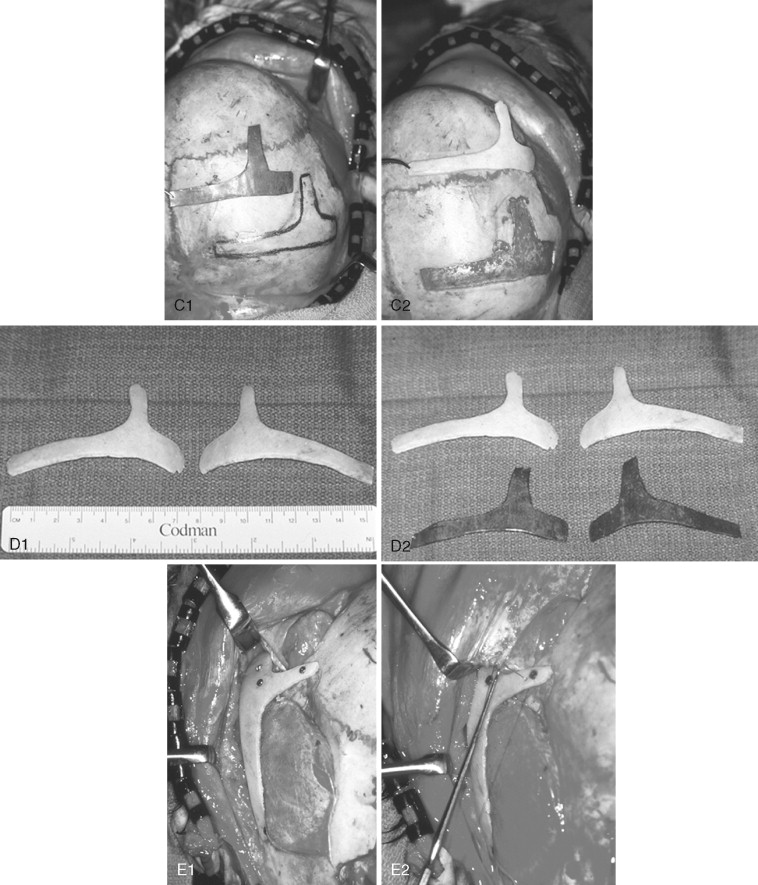
In previously published studies, we have documented the efficacy of reconstructing malar and orbital deficiencies for moderate to severe forms of TCS with the use of full-thickness calvarial bone grafts that have been contoured threedimensionally to form a new zygomatic complex, which then is inset and stabilized with microplate and screw fixation ( Figure 47-7 ). Exposure for the reconstruction is provided by a coronal (scalp) incision without the need for periorbital incisions. Orbital floor defects are reconstructed with fixed split-thickness autogenous cranial bone. Cranial vault graft donor sites are repaired with autogenous split cranial grafts or artificial material. For nonautogenous reconstruction, we prefer to use a titanium mesh base (3D-Mesh; Stryker CMF, Kalamazoo, Mich) fixed to the adjacent skull. The mesh then is filled in with a bone substitute (Bone Source; Stryker CMF, Kalamazoo, Mich). Lateral canthopexies are completed through the coronal (scalp) incision and secured to each new lateral orbital rim. In our study, CT scanning done before surgery and immediately afterward revealed significant increases in lateral orbital wall length, lateral orbital (rim) width, interzygomatic arch width, and zygomatic arch length for patients with TCS. Late (1 year or later) postoperative scans documented that these changes were maintained. No complications occurred during surgery, and repaired skull donor sites healed without clinical defect. Zygomatic and orbital morphologic improvement was achieved in all patients, with follow-up ranging from 24 to 50 months (mean, 35 months) at the close of the study ( Figure 47-8 ).

Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


