Understanding the molecular biology of cancer is an essential step toward developing a rational approach for diagnosis and treatment of the oncology patient. Fundamentally, cancer results from a collection of molecular errors that confer increasingly abnormal proliferative and invasive behaviors. Alterations of regulatory molecules lead to a limited expansion of a single cell until the next mutation occurs and that new mutation, in turn, gives rise to a clonal outgrowth with even greater proliferative potential. Therefore, as the cancer cell “evolves,” it gains the ability to take advantage of and out-compete its normal neighbors. These changes that define the cancer cell are being investigated as valuable targets to better diagnose and treat cancer patients. This chapter provides a broad overview of the types of molecular alterations that govern cancer cell behavior and discusses some of the approaches using these observations as diagnostic and therapeutic targets.
Cancer remains the second leading cause of death in the United States and kills one in five persons in developed countries. Leading causes of cancer include chemical carcinogens, radiation, viruses, age, and genetic background. All five of these risk factors share a common characteristic of altered DNA structure and function. Epidemiologic studies suggest that 80-90% of cancers arise from an accumulation of DNA-damaging events over time. The histopathologic stages observed during tumorigenesis also reflect the time-dependent accumulation of environmental and inherited genetic damage.
Rather than arising de novo , cancers develop through a progression of histologic changes characterized by a breakdown of tissue architecture that is followed by local and distant invasion into surrounding “normal” tissue. For example, squamous cell carcinomas begin as hyperplasia or a thickening of the epithelium ( Figure 33-1, A ). However, cell division is confined to the basal layer of the epithelium and the more superficial layers remain ordered and show differentiation. As tumor progression continues, the epithelium becomes dysplastic. Cell division is observed above the basal layer and the structure of the epithelium loses its order. Dysplasia is further categorized as mild, moderate, and severe based on how superficial cellular atypia occurs within the epithelium. Left alone, dysplasia can remain harmless or can even regress spontaneously. Carcinoma in situ presents with disordered cell division and tissue structure, as well as cells having structural characteristics of cancer cells. However, as the name implies, carcinoma in situ is restricted by the basal membrane and does not invade into the surrounding structures. Squamous carcinoma is characterized by underlying connective tissue invasion. Cancers are further graded by their level of differentiation (well, moderate, or poorly differentiated). Following local invasion of the underlying stroma and surrounding normal structures, the cancer metastasizes.
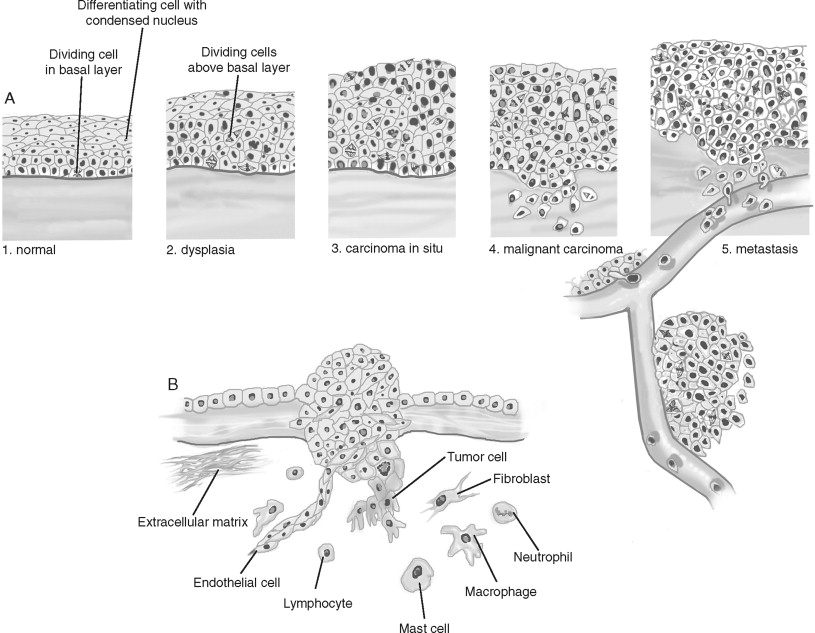
Several important stromal changes occur during cancer progression ( Figure 33-1, B ). The most abundant cell type in the tumor stroma are cancer-associated fibroblasts. Fibroblast proliferation is often referred to as “desmoplasia” and is recognized as a wound response to the cancer. The predominant cell type in this stromal reaction is the myofibroblast, named for the morphologic characteristics of its large, spindle-like shape, indented nuclei, and stress fibers. Angiogenesis is an essential requirement for a tumor mass to grow greater than approximately 1 mm . Only 1 in 600 of these in situ malignancies achieves the blood supply necessary to become a clinically detectable tumor. Rather than invading and appropriating existing vascular beds, tumors recruit and stimulate endothelial cells to construct a tumor-specific blood supply. Lastly, a pronounced inflammatory infiltrate occurs in the stroma. Because immunosuppressed individuals experience elevated levels of certain cancers, it is believed that the immune system surveys and eliminates cancer cells. The innate immune system, consisting of macrophages, dendritic cells, and cytotoxic T-cells/natural killer (NK) cells, detects and kills cancer cells. Monocytes are recruited early and are later differentiated into macrophages. Cytotoxic T-cells recognize and attack cells with tumor-associated antigens (TAAs), releasing factors that inactivate cytotoxic T-cells and recruiting regulatory T-cells (that also inactivate cytotoxic T-cells). However, cancer cells can evade immunosurveillance by down-regulating TAAs. The adaptive immune system consists of T- and B-cells, key stimulators of both the cytotoxic and humoral anti-tumor responses. However, not only can the immune response be suppressed by cancer cells, but some studies indicate that factors produced by immunocytes can support cancer progression. The contribution to and/or inhibition of tumorigenesis by these stromal cell types are active areas of investigation that will be discussed further. In addition to an accumulation and progression of tissue changes, the cancer cell itself shows important alterations from its normal counterparts.
Cancer cells have several structural and behavioral differences from normal cells ( Figure 33-2, A ). Structurally, cancer cells show varying degrees of anaplasia (or degree of differentiation). Both the cytoplasm and nucleus are pleomorphic. The malignant cell size may be either much larger or smaller than its normal counterparts. Typically, cancer cells show a loss of cell adhesion between cells and loss of polarity with respect to the overall tissue architecture. The nuclei of cancer cells typically are disproportionally larger (with respect to the cytoplasm) and denser (or hyperchromatic). Large nucleoli also appear, suggesting high synthetic activity. Normal mitotic spindles are replaced in malignant cells by atypical mitotic figures with tripolar, quadripolar, or multipolar spindles.
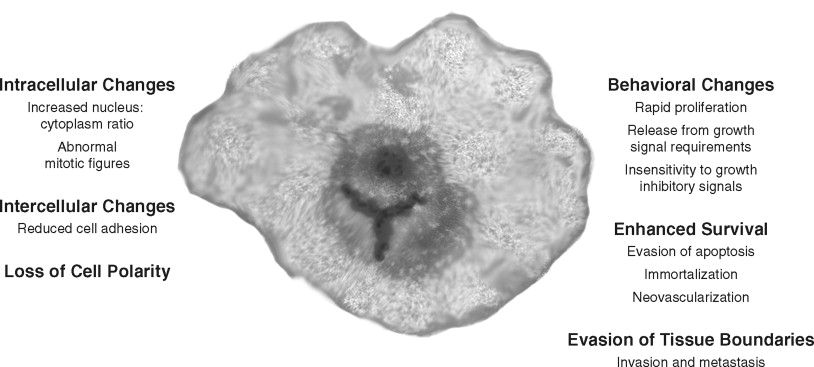
In cell culture and in vivo , cancer cells show key behavioral alterations that are often reflected in the structural changes ( Figure 33-2, B ). Cancer cells often proliferate rapidly and in an uncontrolled manner. In addition, cellular controls leading to apoptosis or cell death are often abrogated. Malignant cells lack the ability to terminally differentiate. Evasion of apoptosis, senescence, and terminal differentiation lead to enhanced cell survival. Most importantly, cancer cells invade the local stroma and induce changes such as angiogenesis. Many malignant cells eventually achieve the ability to metastasize. In summary, several observations suggest that cancer progresses through a series of cellular and histologic changes because of an accumulation of environmentally and hereditarily induced DNA-damaging events.
SIGNAL PATHWAYS ARE ALTERED BY CANCER-ASSOCIATED MOLECULAR CHANGES
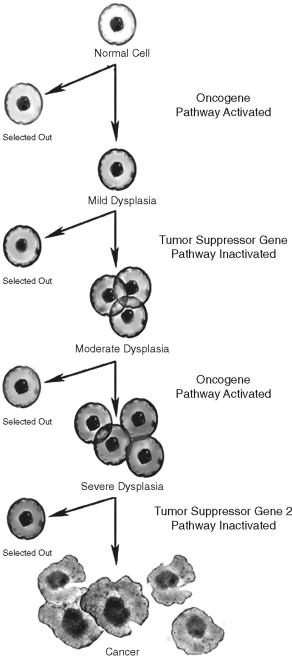
Several lines of evidence suggest that the multiple molecular events leading to cancer development are genetic in nature. In the early twentieth century, Boveri suggested that chromosomal imbalances lead to cancer development. This observation was supported by the study of inherited pediatric tumors and identification of damaged chromosomes in cancer cells. Furthermore, chromosomal DNA damage was linked to cancer formation. DNA repair defects were linked to ultraviolet (UV) light–induced skin cancers. Muller and Auerbach demonstrated the mutagenic potential of ionizing radiation and chemical carcinogens, respectively. Statistical analysis of human tumor incidence and natural history in sporadic and inherited human tumors suggested that 7 to 10 genetic events were required in human tumorigenesis ( Figure 33-3 ). These genetic events were thought to be in two broad categories: oncogenes and tumor suppressor genes.
Oncogenes, or altered growth genes that promote cancer formation (proto-oncogenes), were identified early in molecular investigations in the origins of human cancer. Rous suggested that a retrovirus that caused sarcomas in chickens contained a specific oncogenic sequence. Duesberg and Vogt confirmed that a genetic element induced the chicken sarcoma, and Bishop and Varmus identified the specific oncogene. Weinberg later identified the first human oncogene using non-viral human tumor cells. Because oncogenes could be experimentally added to normal cells to induce a malignant phenotype, oncogenes were initially easier to study.
Tumor suppressor genes, or genetic elements that suppress tumorigenesis, were also postulated early in cancer research. At the cellular level, cancer is a recessive phenotype: the normal cellular activity can suppress malignant transformation. Experimentally, fusion of cancer cells with normal cells resulted in a hybrid cell with normal cell characteristics, suggesting that normal cells had an element(s) that could suppress cancer formation. Later, the addition of specific normal chromosomes to certain cancer cells was found to have the same effect. Using statistical analysis of inherited tumors, Knudson compared the difference between the natural history of inherited versus sporadic cancers and suggested a “two hit” hypothesis in which both copies of a tumor suppressor gene had to be inactivated for cancer formation. Familial cancers, which arose earlier, occurred in patients who inherited a damaged gene copy, thereby reducing the time leading to the inactivation of both copies. This “two hit” hypothesis was supported experimentally after the identification of the first tumor suppressor gene. The retinoblastoma (Rb) gene was cloned by Friend from the pediatric eye tumor (discussed earlier) with a biphasic clinical appearance. In patients who presented early with retinoblastoma (usually in both eyes), a damaged copy of the gene was found to be inherited. In patients with the sporadic form of the disease (usually affecting one eye and presenting later), both inherited copies of the Rb gene were normal. Therefore, in order to understand how cells become malignant, it is important to first understand the nature of gene regulation and how it is altered during human cancer development.
GENE REGULATION OF CELL FUNCTION: A PRIMER
How do genetic alterations lead to cancer cell behavior? Although cellular homeostasis is regulated at many levels, the core of basic cancer research is based on the disruption of the molecular paradigm of gene expression ( Figure 33-4 ). Genes (or coding regions of DNA) are transcribed into messenger RNA (mRNA) that is later transcribed into proteins. The gene products—proteins—govern much of the structure/function of the cell and are the true determinants of the malignant phenotype. Therefore, to understand the role of oncogenes and tumor suppressor genes in cancer biology, it is important to understand its key elements: DNA, mRNA, and protein.
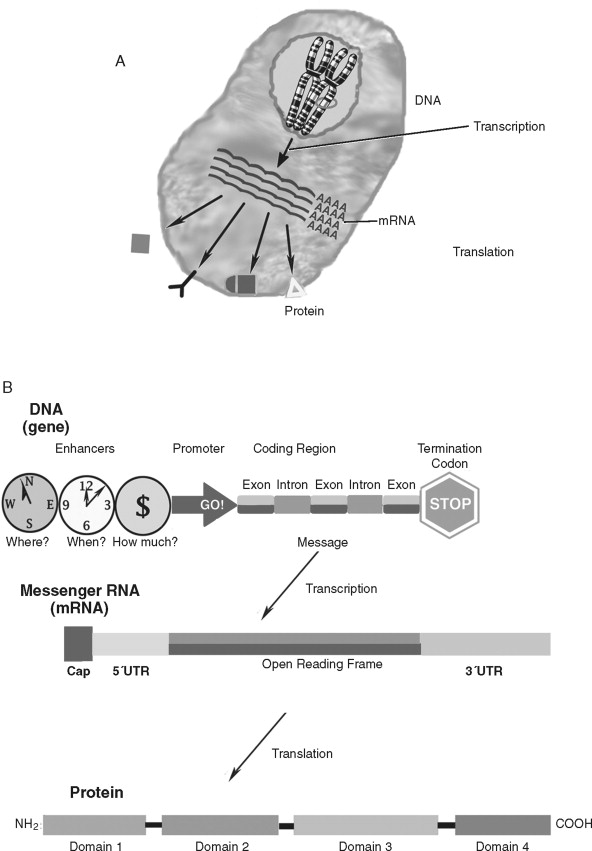
Deoxyribonucleic acid (DNA) is an essential molecule in human health and disease. The 23 pairs of human chromosomes are composed of DNA and protein. Within chromosomal DNA, there are approximately 23,000 to 24,000 paired genes. Therefore, there are two copies of each gene—a DNA region that carries the structural and functional blueprint of the cell. The paired, homologous regions of DNA are called alleles . Genes have three fundamental regions that help govern which sequences are expressed: the enhancer/promoter region, the coding region, and the termination region. The gene promoter/enhancer region, which is bound by transcription factors, determines the activity of a gene. Some genes are continually active; others respond to specific internal/external stimuli. The termination region stops that activity. The coding region contains the message that is transcribed into RNA and subsequently translated into protein. The message translated is based on the order of the nucleotides: adenine, thymine, guanine, and cytosine. The order of these nucleotides is highly regulated. Although there is a redundancy in the code that allows for a limited number of specific changes, nucleotide insertions, deletions, or exchanges can have far-reaching effects (to be discussed later). To prevent nucleotide sequence changes, DNA mismatch repair (MMR) genes provide an important mechanism to protect the integrity of the code.
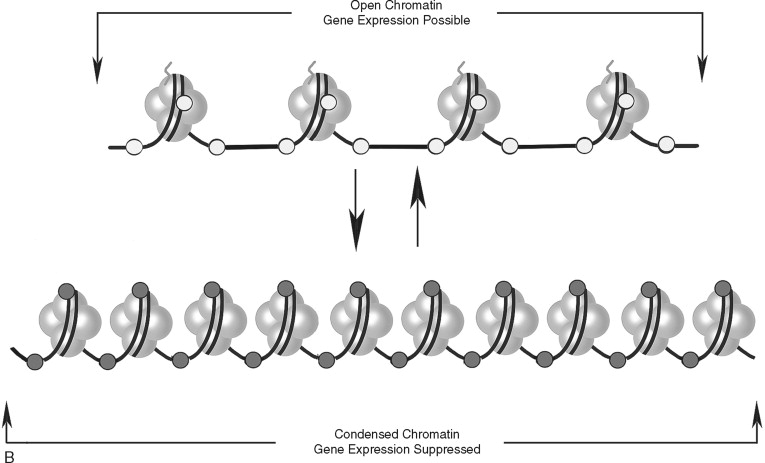
In addition to direct regulation of gene activity, genes are controlled by epigenetic factors. The conformation of chromosomal DNA also regulates the ability of genes to be expressed ( Figure 33-5 ). Chromosomal DNA packaged with histone proteins is called chromatin . Dense chromatin prevents gene expression; open chromatin allows gene activity. The two best understood means of epigenetic control of gene expression are DNA methylation and histone modification. Modification of the nucleotides cytosine and guanine with a methyl group can silence gene activity, whereas removal of the groups can activate gene expression. In addition, modification of histones (a protein family associated with chromosomal DNA) through acetylation, methylation, and phosphorylation is another important control of gene activity. These modifications to DNA and histone change the conformation of the chromosome, leading to dense (low gene expression) and open (high gene expression) chromatin ( Figure 33-5, B ). Therefore, gene regulation is both genetic (or controlled by the promoter/enhancer gene region) and epigenetic (or gene expression control based on chromatin structural conformation near the gene).
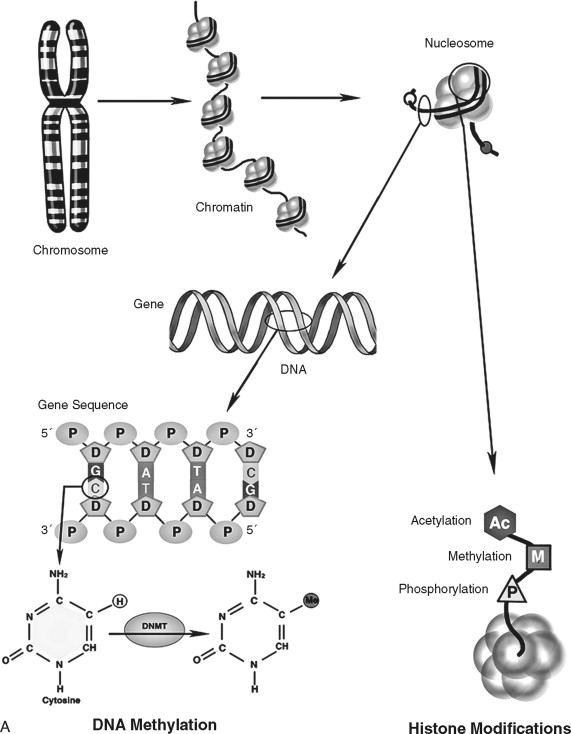
The genetic message encoded in DNA is carried by an intermediary molecule, messenger RNA (ribonucleic acid; mRNA), from the nucleus to the cytoplasm ( Figure 33-4, B ). Outside the nucleus, the message in transcribed into protein. Amounts of mRNA are regulated by two primary mechanisms. First, gene activity and post-transcriptional processing influence how much or how little mRNA is produced. Second, the structure and stability of the mRNA molecule regulates how much transcript is available for protein production. mRNAs have three primary domains. The open reading frame (or region carrying the protein-coding sequence) is flanked by 5′ and 3′ untranslated regions (UTRs). The UTRs play a key role in stability and translation. Many (but not all) mRNAs are further protected by a 5′ cap and poly-adenylated tail that retard transcript degradation. By varying the speed by which mRNA is degraded and/or translated, the cell has another means of regulating itself independently of gene activity. Genes that regulate cell homeostasis (“housekeeping” genes) are often constituently active whereas genes responding to a stimulus, such as proliferation, undergo increases and decreases in cellular levels. Like DNA, mRNA does not affect cell structure or function directly.
Proteins are responsible for many important structural and functional properties of a cell ( Figure 33-4, C ). Each of three nucleotides in the DNA/mRNA coding region is translated into a single amino acid that is added to the protein chain. Proteins often contain several functional groups called domains . For example, the cell surface protein epidermal growth factor receptor contains a ligand-binding domain, a trans-membrane domain, and a tyrosine kinase domain (to stimulate intracellular signaling). In addition to the presence and amount of protein synthesis, protein function is further regulated by chemical and conformational modifications that allow the cell even finer (and more rapid) means of regulation. Proteins can be activated by enzymatic cleavage of inactive precursor molecules. Extracellular proteins are frequently modified by glycosylation, or the addition of complex sugar molecules. Intracellular proteins, especially those associated with signal transduction, undergo the phosphorylation of specific amino acids (serine, threonine, or tyrosine). Acetylation is also a major form of chemical modification of proteins. Environmental changes, such as a change in pH, can profoundly affect protein structure and function. Lastly, protein function can be modulated by feedback inhibition.
Taken together, the constituents of the DNA→mRNA→protein molecular paradigm show a remarkable amount of efficiency and flexibility. Although the cell contains an estimated 23,000-40,000 genes, mRNA and protein processing actually can generate an estimated 1 million separate proteins. Modulation of gene expression exerts a powerful, albeit slower, response to the cell’s needs than protein metabolic and feedback inhibition functions. In addition to structural building blocks, proteins also govern regulatory pathways as ligands, receptors, secondary (or intracellular) messengers, and transcription factors. Proteins act in cascades or pathways. Frequently, proteins producing similar effects within these pathways lead to redundancies that protect and maintain the cellular function governed by the pathway. In short, at each step of the paradigm (DNA→mRNA→protein), cell structure and function can be regulated and modified. Changes in DNA structure and function (gene expression) lead to important qualitative and quantitative protein alterations that ultimately result in the structural and behavioral characteristics of the cancer cell.
NUCLEIC ACID DYSREGULATION IN HUMAN CANCER
Complex biological and biochemical mechanisms protect the integrity of DNA; therefore, there is a low rate of mutagenesis in normal cells. Some genetic diversity must occur to allow for evolution; however, rapid or uncontrolled genomic alterations can lead to cancer or cell death. Because most cancer cells are observed to show at least some DNA alteration, many investigators believe a certain level of genomic instability allows the cancer cell to sustain the necessary genetic damage to lead to the malignant transformation. Variability (in development and in progression) between tumors suggests that tumor cells may differ in their proclivity to accumulate the necessary genetic alterations. Proteins encoded by MMR genes recognize and repair nucleotide errors during DNA replication. Cancer cell genetic alterations can occur as high as 100-fold increase over their non-transformed counterparts. Therefore, in addition to selective pressures on cancer cells, genetic alterations leading to cancer behavior may be due to a failure to survey and repair DNA damage. Genomic instability can be progressive or episodic. Progressive instability results from an inherited defect that leads to new mutations with each successive cell generation. In progressive genomic instability, DNA alterations tend to be widespread in the genome. Episodic instability leads to specific DNA alterations that occur in cells that do not inherit a defect in genomic homeostasis. Rather, episodic instability is triggered by a selective pressure, such as oxidative stress. Both large-scale chromosomal alterations and small-scale nucleotide sequence alterations might result from either type of genomic instability during tumorigenesis. Cancer-associated DNA alterations come in two major groups. First, large-scale chromosomal abnormalities can affect multiple genes and signal pathways. Second, nucleotide sequence alterations affect specific genes. Cancer-associated mRNA changes result from either large- or small-scale DNA alterations, as well as perturbations in other post-transcriptional regulatory mechanisms.
Chromosomal Abnormalities in Cancer Cells
Most human cancers show quantitative (aneuploidy) or qualitative (structural) chromosomal abnormalities. Numerical chromosome changes involve either the entire chromosome or one of the DNA alleles (or pairs). Allelic changes often result in the duplication of the remaining allele. Cells within various tissues tend to share the same numerical chromosomal alterations and can lose a significant number of alleles (25-50%) during malignant transformation. These alterations may be due to progressive instability through inheritance of mutations to DNA damage repair genes such as MMR and p53. Several other events can lead to numerical chromosomal alterations in aneuploid cancer cells. Abnormalities in the number and/or function of the chromosomal centrosome caused by STK 15 kinase or p53 inactivation can lead to aneuploidy. Alteration of mitotic spindle checkpoints, such as hMAD2 in breast cancer, can lead to abnormal segregation of chromosomes during cell division. Lastly, inactivation of genes that respond to DNA damage, such as p53 ATM, BRCA1/2, can also lead to replication of an altered DNA number. In addition to numerical chromosomal alterations, three forms of chromosomal structural alteration may be critical for malignant progression: gene amplification, rearrangement/translocation, and deletion.
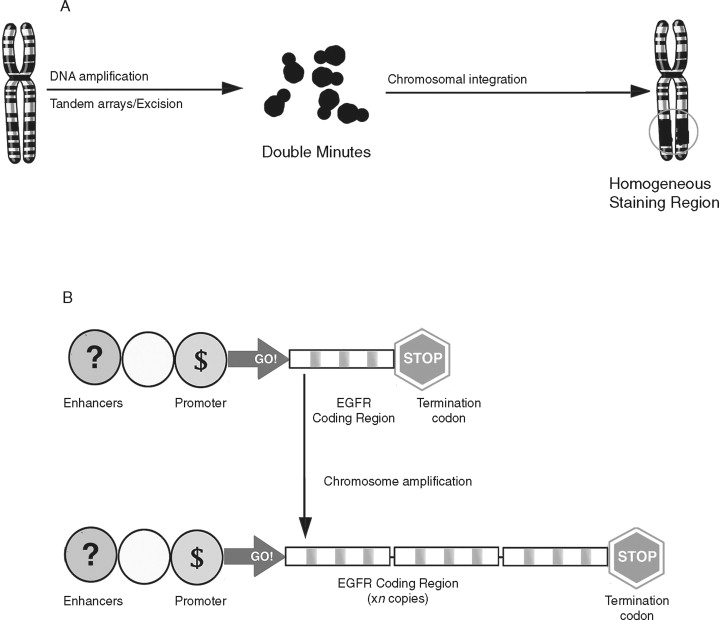
Gene amplification is the repeated duplication of a certain chromosomal region, often 50-100–fold. ( Figure 33-6 ). There are two distinct structural alterations that lead to amplification. Double minutes are small pieces of chromosome that lack a centromere. They cannot be inherited but are maintained by selective pressures. Homogeneous staining regions are double minutes that are incorporated into the genome and are inherited. Increased gene copy number can lead to overexpression of that gene. Amplification is believed to occur late in tumor development and has been linked with resistance to chemotherapeutic agents. Both double minutes and homogeneous staining regions are frequently observed in chemotherapy-resistant tumors and are believed to harbor genes that confer a selective advantage to the cancer cell.
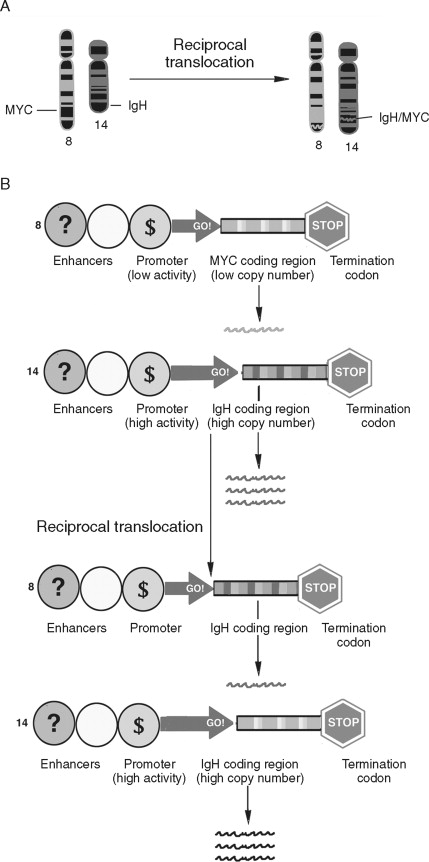
Chromosomal rearrangements such as translocations have been well described in human cancers ( Figure 33-7 ). Translocations are a transfer of part of one chromosome to another. Translocations are considered reciprocal if two chromosomes trade material. Like gene amplification, translocations can lead to cancer-associated alterations of gene expression. In Burkitt’s lymphoma reciprocal translocation of the Myc proto-oncogene to an immunoglobulin promotor in B-cells leads to an elevated expression of Myc. Therefore, unlike gene amplification, which increases gene expression by increasing template number, this example of reciprocal translocation leads to increased gene expression by changing the transcriptional regulatory apparatus. The Philadelphia chromosome (a translocation from chromosomes 9 to chromosome 22) is an important marker for chronic myelogenous leukemia. Rather than alter the expression level of the genes at this locus, a fusion gene product, BCR-ABL, results. Activation of both oncoproteins is important to tumor progression.
Chromosomal deletions are believed to be an important mechanism of inherited loss of tumor suppressor gene loci ( Figure 33-8 ). Following the sporadic or inherited deletion of one allele, the second can be inactivated by either mutation or epigenetic “silencing” (which is discussed later) in a “two hit” fashion. In colorectal cancer, deletion of important suppressor gene loci at chromosome 5 (APC), 17 (p53), and 18 (DCC) are important molecular events leading to malignant progression. Whereas translocations and deletions are often linked to genes that promote malignant transformation, chromosomal deletions are primarily associated with loss of genes that suppress cancer formation.
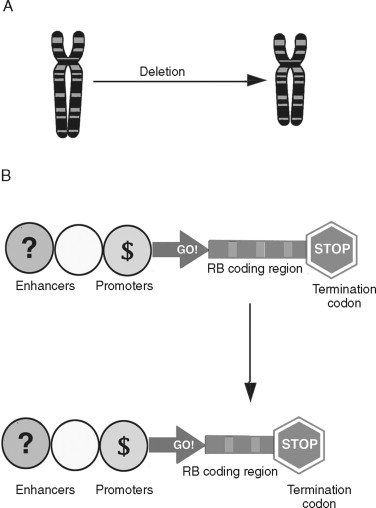
Nucleotide Sequence Abnormalities in Cancer Cells
Rather than large-scale changes in chromosomal DNA, nucleotide sequence alterations affect individual genes. These alterations usually involve a single nucleotide change or insertion/deletion of nucleotides ( Figure 33-9 ). Changes in a single nucleotide (point mutation) can lead to a missense mutation (changing an amino acid) or nonsense mutation (stopping protein synthesis). Inserting or deleting nucleotides are classified as frameshift mutations because the numerical change in nucleotides shifts how the message is read, leading to a completely altered amino acid sequence. This altered amino acid sequence can lead to profound changes in protein function. A common nucleotide alteration observed in cancer cells is microsatellite instability (MSI).
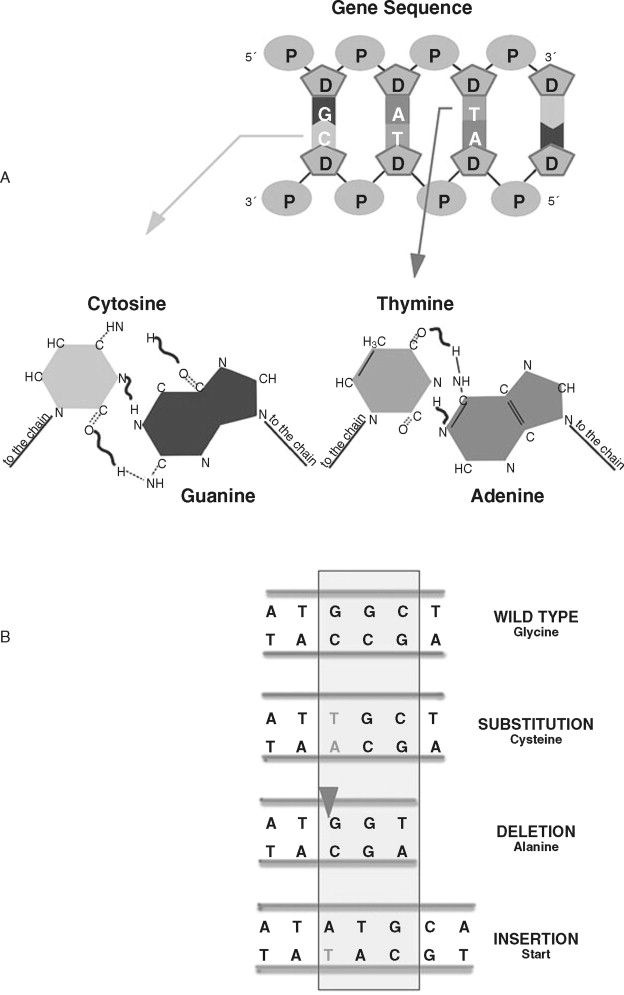
MSI in cancer cells refers to insertion or deletion of nucleotides in simple repeated sequences that are normally found in DNA. Microsatellites are repeats of four (or more) nucleotides and appear as numerous and randomly distributed regions throughout the chromosomal DNA. The frequency of microsatellite alteration depends, in part, on the type of repeated sequence (mononucleotide repeat, dinucleotide repeat), length (larger alleles are more frequently altered), location of the sequence, and the type of alteration in DNA repair. Alteration of MMR genes such as hMSH1/2/3, hPMS1/2, and hMLH3 is associated with an increased frequency of MSI. MMR-deficient cancer calls can show an approximately 100-fold increase in MSI over MMR-proficient cancer cells. Microsatellite alteration leads to frameshift mutations in important proteins regulating normal cellular homeostasis, such as TGFβRII, BAX, APC, IGFIIR, and others.
Epigenetic Alteration of Gene Expression in Cancer Cells
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


