Species
Mass (%)
H3PO4
48.8
Al
1.6
Zn
6.1
H2O
43.3
The sensitivity of these properties to acid concentration led to practical difficulties in the deployment of dental silicate cements. The optimum concentration of phosphoric acid is stable only in an atmosphere of 70 % relative humidity. In more humid atmospheres, the solution takes up water, reducing the acid concentration and leading to inferior cements (Paffenbarger et al. 1938; Wilson et al. 1970a; Worner and Docking 1958). Similarly, in less humid atmospheres, the acid liquid loses water and increases the concentration of the acid. Cements made from such solutions are also inferior. This sensitivity to the prevailing humidity was a further disadvantage of these cements as practical dental materials.
1.2.2 The Setting Reaction of Dental Silicate Cements
A critical step in the development of the glass-ionomer cement was the understanding of the setting reaction of the dental silicate cement, which was achieved by Wilson et al. from 1968. The earliest view had been that the cement was formed by gelation of silicic acid in the solid state (Ray 1934). This was shown to be incorrect in a study of the effect of acid storage environments on dental silicates, from which Wilson and Batchelor concluded that the matrix could not be silica gel (Wilson and Batchelor 1968). They suggested initially that the matrix might be a silico-phosphate material instead, but it was then found that infrared spectroscopy could not detect the presence of any P–O–Si bonds (Wilson and Mesley 1968). The nature of the setting reaction was finally established in 1970 in a paper by Wilson et al. (1970b) which, while primarily concerned with the role of water in these cements, showed that the main products of setting were amorphous aluminium phosphates. Some siliceous material was formed, but it remained as a coating around the partly reacted glass filler particles. The set cement was thus shown to consist of partly reacted glass particles embedded in a matrix of calcium phosphate.
Careful examination of the results obtained led Wilson to conclude that the setting of the dental silicate cement took place as follows. As the powder and liquid are mixed, the reaction begins with the attack on the basic glass particles by hydrogen ions from the phosphoric acid solution (Wilson and Mesley 1968; Wilson and Batchelor 1967a). This causes migration of aluminium, calcium and sodium ions into the matrix (Wilson and Kent 1968) and leaves behind what is essentially silicic acid. Fluoride ions are also released into the matrix at this stage (Wilson and Kent 1968), possibly as complexes of the type AlF2+ and AlF2 +, both of which are known from other studies (Connick and Poulsen 1957; O’Reilly 1960; Akitt 1989).
As the metal ions migrate into the matrix phase, the ionised acid forms metal salts based on the anionic species H2PO4 − (Kent and Wilson 1969). As this happens, the pH of the matrix rises towards neutral and the reaction gradually ceases. The products formed are substantially insoluble and also rigid, and their formation thus causes the cement to set to a hard non-deformable body. Wilson et al. referred to this process as “precipitation”, a term that is not wholly satisfactory as precipitation strictly refers to the formation of an insoluble solid that separates from the solution in which it is formed. Dental silicate cements, like other materials of this type, show no phase separation. Instead, all of the water originally present in the acid solution becomes incorporated into the final cement. In fact, one of the defining features of cements is that they consist of pastes which set hard in their entirety, without expelling water (Wilson and Nicholson 1993).
Despite this infelicity in terminology, the studies by Wilson et al. offered a brilliant explanation of the setting of dental silicate cements. Their results showed that hardening reached 65 % of its final value within 30 min and ceased after about 72 h (Wilson et al. 1972). The pH was shown to reach 5.2 after 48 h. However, electrical conductivity studies showed that some form of reaction continued slowly for at least 7 weeks (Wilson and Kent 1968), though hardness did not change. Strength had been known for some years to continue rising in these materials for at least 12 months after fabrication (Paffenbarger et al. 1938), and hence, the discovery of a slow long-term reaction in the solid state was not particularly surprising. The final matrix, as shown by electron probe microanalysis (Kent et al. 1970), is predominantly amorphous aluminium phosphate that also contains fluorite (CaF2) and sodium acid phosphates.
As previously mentioned, silicic acid is also formed in the early stages of the setting process. This substance polymerises readily to form silica, SiO2 (Kent et al. 1970; Tarutani 1989). This result is consistent with the known aqueous chemistry of silica (Iler 1979), in particular that low pH favours the occurrence of silicic acid, Si(OH)4, but this changes rapidly in the pH range 5–6, and gelation to silica becomes favoured (Iler 1979).
Water also plays an important role in the setting and post-hardening reactions of these cements. Its initial function is as the medium for the reaction, but as the setting proceeds, it hydrates the products of reaction and becomes incorporated into the solidifying mass. Studies have shown that water exists in fully hardened cements in two states, namely, bound and unbound water. These have been named alternatively as non-evaporable and evaporable, respectively (Wilson et al. 1979). These two states differ in that bound water is retained by the cement when stored in a strongly desiccating atmosphere for 24 h, whereas the unbound water is lost under these conditions, causing a reduction in mass (Wilson et al. 1979). The precise location of the bound water is not clear, though the possibility of hydrating the components of the aluminium phosphate, particularly the Al3+ ions, is likely (Enderby and Nielson 1989).
In their studies of the set cement, Kent et al. 1970 (Tarutani 1989) used glass powders that had been sieved to remove the finest particles. Electron probe microanalysis showed that the matrix of the dental silicate cement contained Al, P, Na and F only, with the Si restricted to layers around the partly reacted glass filler particles. This was important in establishing how these materials set, but it is not a true picture of the elemental distribution in clinical dental silicates. In a later study, Brune and Smith (1982) showed that silicon was distributed throughout the matrix. It seems probable that this was caused by the reaction of very fine glass particles that were so small that they degraded completely as the reaction proceeded. Hence, this later observation does not invalidate the conclusions of Wilson et al. (1970b) that the setting of dental silicates does not consist of the formation of silicates but of phosphates.
1.2.3 Physical Properties of Dental Silicate Cements
Physical properties of dental silicates vary with the powder/liquid ratio and the best cements are mixed at high ratios, up to 4 g/cm3 (Wilson et al. 1972). Mixing was done by gradually incorporating the powder, so as to minimise the effect of the reaction exotherm between the components of the cement (Brune and Smith 1982). Properties of the resulting cements are shown in Table 1.2.
Table 1.2
Properties of clinical dental silicate cements
|
Property
|
Value
|
|---|---|
|
Powder/liquid ratio
|
2.70–4.02
|
|
Working time (23 °C), min
|
3.6
|
|
Setting time (37 °C), min
|
3.25–7.0
|
|
Compressive strength (24 h), MPa
|
68.5–255
|
|
Flexural strength (24 h), MPa
|
24.5
|
|
Tensile strength (24 h), MPa
|
13.6
|
|
Solubility/disintegration strength (24 h), %
|
0.34–0.38
|
|
Opacity, C0.7
|
0.42–0.71
|
The values of strength are all quoted at 24 h, but they all continue to rise for at least a year after fabrication (Paffenbarger et al. 1933). The overall strength of the dental silicate is generally considered to be higher than that of any other acid–base cement, though glass-ionomers with strengths exceeding 300 MPa in compression have been reported (Guggenberger et al. 1998).
1.2.4 Solubility and Ion Release
The properties of dissolution and ion release from dental silicate cements were widely investigated, as they were of such importance in the clinical performance of these materials. Erosion and dissolution limited the acceptability of dental silicates, but fluoride release was beneficial in view of the therapeutic effect of fluoride for teeth damaged by caries (ten Cate et al. 2008).
When fully set, dental silicate cements were resistant to attack in neutral solutions. The main species eluted were sodium, fluoride and silica, all in very small amounts (Wilson and Batchelor 1967a). However, before hardening was complete, the cements were vulnerable to attack even in neutral conditions because of the presence of a variety of water-soluble intermediates, including sodium salts, acid phosphates and fluorides.
The removal of such matrix-forming ionic species was found to cause significant changes to the cement surface. To prevent this, clinicians adopted the practice of covering the surface of a freshly placed dental silicate with a layer of varnish. Once a properly formulated dental silicate had hardened fully, it was safe from attack by neutral solutions. It should be noted, though, that poorly formulated cements, including those prepared from incorrect powder/liquid ratios, could contain relatively large amounts of soluble reaction products and consequently disintegrate when immersed in neutral solutions. Studies showed that the main species eluted into neutral solutions were soluble salts of sodium and phosphate and also fluoride (Wilson and Batchelor 1967a, b). As time passed, the rate of elution dropped rapidly, and acid phosphate species (H2PO4 − and HPO4 2−) ceased to be released.
By contrast, in acidic conditions, erosion of these cements was shown to be much more rapid (Wilson and Batchelor 1968). This susceptibility to acid attack was suspected as being the cause of the variable performance of these materials from the beginning (Voelker 1916), a view that was confirmed during the 1950s (Norman et al. 1957) and 1960s (Jorgensen 1963). The extent of erosion by acids was found to vary not just with pH but also with their complexing ability. Citric acid, in particular, was found to be especially erosive (Wilson and Batchelor 1968; Stralfors and Eriksson 1969), a concern given the extent to which citric acid drinks are consumed. Studies of erosion of dental silicate cements in service showed that they tended to develop a pattern of grooving at the margin of the restoration and the tooth (Tay et al. 1974, 1979) and, when left unchecked, led to eventual failure of the restoration.
Although dental silicate cements failed under acid attack, their resistance was compared favourably with all other dental cements except the glass-ionomer (Norman et al. 1959; Kuhn et al. 1984). This lack of acid resistance was not considered a particular problem; in fact, when properly prepared and placed, dental silicate cements gave very good results in patients (Robinson 1971). The problem was that the range of optimum performance was relatively narrow. Poor handling and preparation, allowing the acid concentration to change by exposure to the atmosphere at varying humidities and preparing the cement at less than the recommended powder/liquid ratio were all factors that would lead to a cement containing too much water-soluble material and result in clinical failure. It was this unreliability that gave the dental silicate cement its reputation as an unsatisfactory material and left the door open for the development of a much less sensitive material: the glass-ionomer cement.
1.3 Zinc Polycarboxylate Cements
The development of the zinc polycarboxylate cement was also an important step in the invention of the glass-ionomer cement. First reported by Smith in 1968 (Smith 1968), this cement was prepared from the reaction of zinc oxide with aqueous poly(acrylic acid). The setting process involved gelation of the poly(acrylic acid) solution with zinc ions released from the powder as a result of attack by the acid. Setting involved incorporation of all of the water in the original acid solution and the formation of a rigid mass from the initial viscous paste.
The zinc polycarboxylate cement was invented as a result of a rational exploration by Smith of factors that can cause adhesion to the tooth surface, and the work was carried out with the aim of producing a material that could act as a luting cement yet show true adhesion to the tooth (Smith 1968). It was the first inherently adhesive dental material and, as such, represented an important advance in restorative dentistry.
The zinc polycarboxylate cement remains a useful material for clinical dentistry, with applications as liners and bases, luting cements and periodontal packs (Wilson and Nicholson 1993). It can be used in all applications of zinc phosphate cements, with the possible exception of post crowns and cantilever bridges (Smith 1982). This does not mean that zinc phosphate has become obsolete or completely superseded in these applications. The choice of material varies between clinicians, and individual clinical technique may favour one material over the other. Consequently, zinc phosphate is still employed in the restorative dentist’s armamentarium.
The critical aspect of the invention of the zinc polycarboxylate was the use of aqueous solutions of poly(acrylic acid) as the cement-forming liquid. Concentrations were relatively high, typically 30–43 % by mass (Wilson and Nicholson 1993), which means that the liquid had a reasonably high viscosity. Although poly(acrylic acid) is the principal acid polymer used in these cements, other polymers were quickly proposed, including copolymers of itaconic or maleic acid (Bertenshaw and Combe 1972).
The molar mass of the polymers used varies between about 22,000 and 49,000 (Bertenshaw and Combe 1976) and needs to be carefully controlled in order to obtain satisfactory cements. High molar mass polymers lead to cements of high strength; however, the consequently high viscosity of the polymer solution makes mixing the cement difficult. As a result, molar mass tends to represent a balance between these conflicting requirements and is chosen as a compromise between being low enough to allow satisfactory mixing and high enough for useful strength. Similar issues arise with the glass-ionomer cement, and molar mass of the constituent polymer for these materials also represents a compromise between the need for ease of mixing and high final strength.
The powders for zinc polycarboxylate cements are also complex and do not consist simply of pure zinc oxide (see Table 1.3). As is the case for zinc phosphate cements, the zinc oxide for zinc polycarboxylates has to be deactivated for use in practical cements. Pure powdered zinc oxide reacts too quickly with aqueous poly(acrylic acid) solution to form a smooth paste. Instead, it forms clumps of powder agglomerated by prematurely formed zinc polyacrylate, and this lumpy mixture cannot be mixed further to form a smooth usable paste.
Table 1.3
Composition of zinc polycarboxylate cements
|
Component
|
% by mass
|
|---|---|
|
Zinc oxide powder: ZnO
|
85.2–96.8
|
|
MgO
|
4.73–10.06
|
|
Poly(acrylic acid), concentration in solution
|
32.4–42.9
|
Zinc oxide is deactivated partly by sintering, as mentioned previously for zinc phosphate, a process which results in a pale yellow powder that is very slightly oxygen-deficient (Dollimore and Spooner 1971). In addition, the zinc oxide is mixed with up to 10 % by mass of magnesium oxide powder. Powders may, in addition, contain silica, alumina or bismuth salts, the latter to impart radiopacity. Another key additive is stannous fluoride, which is added at 4–5 % by mass, and was originally added as a source of fluoride (Foster et al. 1974). In fact, stannous fluoride itself probably dissolves out of these cements, rather than free fluoride, as SnF2 is soluble in water and exists in aqueous solution at very low levels of dissociation (Turner et al. 2013). In addition to providing a source of fluoride, stannous fluoride was found to increase the strength of zinc polycarboxylate cements (Foster et al. 1974), a phenomenon that has yet to be explained.
The majority of brands of zinc polycarboxylate cement consist of two components, an appropriately formulated deactivated zinc oxide powder and an aqueous solution of polymeric acid, typically poly(acrylic acid). Quite early on, it was found that this cement could be formulated as a dry powder consisting of the zinc oxide powder plus dried polymer, with reaction initiated by mixing this powder with the correct volume of pure water (Bertenshaw and Combe 1972). Setting of these water-activated cements proceeds satisfactorily, and there appear to be no important differences in setting, ultimate strength or clinical performance between these materials and cements formulated more conventionally from aqueous acid solutions.
The way in which zinc polycarboxylate cements are formulated has been shown to influence cement properties, including working and setting times, and strength when set. Factors affecting these include powder/liquid ratio, composition of the powder, molar mass and type of the polymeric acid and also its concentration in the cement-forming liquid (Smith 1971). Typical properties of these cements are shown in Table 1.4.
Table 1.4
Properties of zinc polycarboxylate cements
|
Property
|
Value
|
|---|---|
|
Working time (23 °C)/min
|
2–5
|
|
Setting time (37 °C)/min
|
3–12
|
|
Compressive strength (24 h)/MPa
|
48–80
|
|
Tensile strength (24 h)/MPa
|
4.8–15.5
|
|
Adhesion to enamel (tensile, 24 h)/MPa
|
4.1–6.9
|
|
Adhesion to dentine (tensile, 24 h)/MPa
|
2.2–5.1
|
Setting and hardening of zinc polycarboxylates occur reasonably quickly. A typical formulation reaches its maximum strength at 24 h, after which it shows little or no change over subsequent time periods (Watts et al. 1979; Osborne et al. 1978; Paddon and Wilson 1976). The cement shows some viscoelastic properties, and measured strength is influenced by the crosshead speed of the testing machine, particularly at low speeds (Wilson and Lewis 1980). This viscoelasticity remains as zinc polycarboxylate cements age, and distinct creep has been detected under static loading conditions in samples aged for 24 h (Wilson and Lewis 1980). Significant stress relaxation was also found in specimens aged for 4 weeks (Paddon and Wilson 1976).
Zinc polycarboxylates were the first adhesive dental restorative materials and have been shown to bond to untreated dentine and enamel (Mizrahi and Smith 1969). Bond strength to enamel is higher than to dentine, as shown in Table 1.4. In general, zinc polycarboxylate appears to be mild in clinical use, having minimal effects on the dental pulp (Plant 1970; Beagrie et al. 1972; Wilson 1968). They are also nonirritating when used in implants in soft tissue and bone (Wilson 1968).
1.4 Invention of the Glass-Ionomer Cement
It is important to understand the nature of the invention of the glass-ionomer cement. It did not consist of simply mixing the glass powder of the dental silicate cement with the poly(acrylic acid) solution of the zinc polycarboxylate and finding that an excellent cement was the result (Wilson 1996a). It is true that early attempts to improve the dental silicate cement did involve experiments with aqueous solutions of organic acids, though these acids were monomeric and the results, while interesting, were not successful in producing cements with significantly improved properties (Wilson 1968).
Poly(acrylic acid) was considered in those early experiments, but the results were so disappointing that they were not reported at the time (Wilson 1968). Instead, they were hinted at some years later (Wilson and McLean 1988) and not described in detail until Wilson published his personal account of the invention of glass-ionomers (Wilson 1996a). It turned out that these early glass-polyacrylate cements were disappointing in the extreme. They formed an intractable paste which underwent a sluggish reaction, as observed by an increase in the viscosity of the paste; hardening was very slow and the product was hydrolytically unstable. This cement was clearly of no use as a potential dental material (Wilson 1996a).
The invention of the glass-ionomer cement was, in fact, not a single act but rather a series of innovative steps. One key step was the finding that the alumina/silica ratio of the glass controlled the resulting basicity and hence the readiness with which a glass powder would react with an acid solution. This pointed the way towards the first successful glass for a glass-ionomer cement, one with sufficient basicity to react with aqueous poly(acrylic acid). Controlling the alumina/silica ratio allowed the glasses to set much more rapidly than the original glass-poly(acrylic acid) mixture, where the glass was of relatively low basicity. These improved mixtures not only set more rapidly; they were also hydrolytically stable when placed in water (Wilson and Kent 1971; Kent et al. 1973).
The first glass to give at least moderately satisfactory cements was designated G200 and contained alumina and silica in the appropriate ratio to give a high basicity; it was also high in fluoride. Its composition is given in Table 1.5. The resulting cement was known as ASPA 1, the term ASPA being an acronym for aluminosilicate poly(acrylic acid). ASPA was also the brand name of the very first commercial glass-ionomer cement, launched in 1975 (Wilson and McLean 1988).
Table 1.5
Composition of the glass G200
|
Component
|
% by mass
|
|---|---|
|
SiO2
|
30.1
|
|
Al2O3
|
19.9
|
|
AlF3
|
2.6
|
|
CaF2
|
34.5
|
|
NaF
|
3.7
|
|
AlPO4
|
10.0
|
The first glass-ionomer, based on G200, was far from the finished article, in that it set relatively slowly and retained a high degree of water sensitivity for a considerable time after setting, which was a distinct drawback. In addition, the glass itself was not particularly translucent, and therefore, the resulting cement had poor aesthetics.
The next inventive step along the path to creating a satisfactory glass-ionomer cement was the discovery of the effect of (+)-tartaric acid on the setting reaction. This discovery came about from a consideration of the nature of the G200 glass. It was exceptionally high in fluoride, a factor that seemed to be important as G200 was the only glass capable of forming a usable cement with poly(acrylic acid) solution (Wilson 1996a). Wilson and his team inferred that fluoride ions must play an important part in controlling the setting reaction, suggesting in particular that they interacted with aluminium ions released from the glass and prevented them from prematurely cross-linking the poly(acrylate) polymer chains. This, they postulated, was because of the high affinity of aluminium for fluoride ions and the formation of complex ions of the type AlF2+ and AlF2 +, which had been proposed to occur during the setting of dental silicate cements (Wilson and Kent 1968).
Slowing the reaction of aluminium via chelation seemed to be a way forward. In the gravimetric analysis of rocks, the established method to prevent premature precipitation of aluminium as the phosphate was to include either citric acid or (+)-tartaric acid, both of which were known to form water-soluble complex ions with Al3+ (Lundall and Hoffman 1938). These two acids were therefore examined as potential additives in the glass-ionomer cement.
The results were striking, especially with (+)-tartaric acid. The resulting cements had longer working times and sharper setting, seemingly contradictory properties, both of which made the cement mixture easier to mix and to manipulate (Crisp et al. 1975; Wilson et al. 1976). The cements also had improved compressive strength and were more resistant to acid attack. Wilson considered this to be the most important discovery made in the whole process of inventing and developing the glass-ionomer cement (Wilson 1996a). The new formulation was named ASPA II, and it can be considered to be the first really practical glass-ionomer cement.
1.5 Pioneering Studies of Glass-Ionomer Cements
Following the preliminary reports announcing the development of the glass-ionomer cement (Wilson and Kent 1971; Kent et al. 1973), detailed scientific publications on these cements began to appear in 1974. As well as scientific studies, the first clinical report appeared that year (McLean and Wilson 1974). It covered fissure sealing and restoration with glass-ionomers and was the report of a 2-year clinical study undertaken with the original ASPA formulation of glass-ionomer.
The initial scientific studies were concerned with the setting reaction of these materials (Crisp and Wilson 1974a, b; Crisp et al. 1974). They broke the reaction down into two stages: the decomposition of the glass powder (Crisp and Wilson 1974a) and the reaction of the ions released to cross-link the polyacid, which was incorrectly termed “precipitation” (Crisp and Wilson 1974b). Gelation would have been a better term for this part of the setting process.
The remaining paper in this important series describes the application of infrared spectroscopy to the study of the setting reaction (Crisp et al. 1974) and was important in showing that the main products are calcium and aluminium polyacrylates. These two possible products can be distinguished on the basis of their infrared spectra. Calcium polyacrylate shows a carboxylate band at 1540 cm−1, indicating a highly ionic structure, whereas aluminium polyacrylate shows an equivalent band at about 1600 cm−1, indicating a degree of covalent character with distinct chelation of the central Al3+ ions by the surrounding carboxylate groups (Crisp et al. 1974). This paper also showed that the fully set cement contained some residual unreacted carboxylic acid groups, trapped within the cement as it hardened and unable to react for steric reasons (Crisp et al. 1974).
1.6 Early Research on Glass-Ionomers
1.6.1 The Composition and Structure of the Glasses
The first practical glasses for use in glass-ionomer cements were calcium aluminosilicates with added fluoride (Wilson and Kent 1971; Kent et al. 1979; Wilson et al. 1980). Their alumina/silica ratio was adjusted to make them sufficiently basic to set on reaction with aqueous poly(acrylic acid). Although numerous other glass systems have been investigated since these early studies, the glasses developed by Wilson and Kent remain the basis of all practical glass-ionomer cements used clinically. The one substantial change has been the development of strontium-containing ionomer glasses (Guida et al. 2003), where the element strontium is used in place of calcium in the formulation.
Fluoride is an essential component of the glasses (Wilson and McLean 1988). This element has several functions within the glass. It lowers the fusion temperature, it improves the working behaviour of the freshly mixed cement paste by preventing premature gelation and it improves the strength of the set cement. Fluoride-containing glasses are much less opaque than pure oxide ones, and this in turn means that cements prepared from them have improved translucency.
The essential property of glasses for use in glass-ionomer cements is that they are basic and can react with aqueous solutions of acid (Hill and Wilson 1988). This property arises from the ratio of alumina to silica in the glass. Right from the start of research into workable glasses for these cements, this need for basicity and how it could be achieved were well understood. The theoretical framework for this understanding was the random network model of glass as advanced by Zachariasen in 1932 (Zachariasen 1932).
This model considers the glass structure to be a random assembly of oxygen polyhedral, each comprising a small central cation surrounded by a number of negatively charged oxygen ions. A typical polyhedron is (SiO4). These structures are linked at the corners via the oxygen ions to form an array of chains and interconnected units. The overall concept thus views a glass as consisting essentially of a polymer based on (SiO4) tetrahedra joined at the corners and exhibiting varying degrees of cross-linking.
The reactivity of such glass structures towards aqueous acids can be increased by including cations such as calcium that can break up the continuous Si–O–Si structural units to form non-bridging oxygen. These ions are described as “network modifying”:
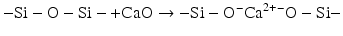
The inclusion of aluminium has more complex effects than the inclusion of simpler chemical species such as calcium or sodium. Aluminium can act as a network modifier in an analogous way to calcium, but it can also be a network former. In the latter case, the presence of reasonable amounts of silica as tetrahedral building blocks of (SiO4) forces the aluminium to adopt a similar tetrahedral geometry and to form species equivalent to (AlO4). Although these tetrahedra are the same size as the (SiO4) ones, they carry more formal charge. This is because the central cation is a 3+ ion, compared to the formal 4+ charge on the central silicon in (SiO4). The presence of the (AlO4) tetrahedra therefore has to be balanced by additional cations (e.g. Na+, Ca2+) in the structure close to the main oxide network (Lowenstein 1954).
These ideas show that the aluminosilicate glass structure of ionomer glasses can be regarded as consisting of linked (SiO4) and (AlO4) tetrahedral, with additional cations to balance the charge deficiency due to aluminium. It is the alumina units and their associated cations that serve as the point of reaction with aqueous acid solution, and reaction of the acid involves removal of the charge-balancing cations, followed by rupture of the aluminosilicate network (Wilson and McLean 1988). The Al/Si ratio cannot apparently be greater than 1:1; otherwise there are insufficient (SiO4) tetrahedral to force the aluminium into fourfold co-ordination, and high basicity does not develop (Lowenstein 1954). Glasses for ionomer cements must have a minimum Al/Si ratio of 1:2, with practical glasses having ratios above this limit (Kent et al. 1979; Wilson et al. 1980). The upper limit in this ratio was found to be 0.75:1 by mass early in these studies (Wilson and McLean 1988), this limit being the point at which the mineral phase corundum (Al2O3) crystallises out within the glass structure. Glasses containing two distinct phases are inherently opaque, and resulting cements lack the aesthetics for clinical use.
The three essential components of the early glasses for ionomer cements were silica, alumina and fluorite, i.e. SiO2, Al2O3 and CaF2. In practice, additional components were added in order to improve properties such as the setting rate, translucency and final strength (Wilson and McLean 1988). Practical glasses typically belonged to complex systems such as:
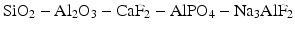
The glasses were prepared by fusion of these components, typically in a ceramic crucible, with fusion temperatures varying between 1100 °C and 1500 °C, depending on the precise chemical composition of the fusion mixture (Wilson and McLean 1988; Hill and Wilson 1988). After melting and allowing the mixture to become thoroughly homogeneous at an elevated temperature, the melt was cooled rapidly by pouring it either onto a metal plate or directly into water. This resulted in the formation of a glass frit consisting of large pieces of glass. It was then ground to a fine powder, typically of 20–50 μm, depending on the clinical application of the cement (Wilson and McLean 1988).
The preparation results in glasses of varying structures. Some show a degree of phase separation that leads to an opaque appearance (Hill and Wilson 1988), whereas others have no visible phase separation and are clear in appearance. Phase-separated glasses were found to give rise to stronger cements than clear glasses (Wilson and Nicholson 1993; Kent et al. 1979), as shown by the results in Table 1.6.
Table 1.6
Relationship between composition and cement properties for early ionomer glasses
|
Code
|
G241
|
G278
|
G279
|
G282
|
|---|---|---|---|---|
|
Composition
|
||||
|
SiO2
|
120
|
120
|
120
|
120
|
|
Al2O3
|
102
|
102
|
102
|
102
|
|
CaO
|
112
|
101
|
84
|
11.2
|
|
CaF2
|
0
|
15.6
|
39
|
140
|
|
Appearance below T g
|
Clear
|
Clear
|
Clear
|
Opaque
|
|
Powder/liquid ratio/g cm−3
|
2.5
|
2.5
|
2.0
|
2.5
|
|
Setting time (37 °C)/min
|
2.25
|
2.25
|
a
|
3.0
|
|
Compressive strength (24 h)/MPa
|
74
|
125
|
a
|
165
|
The structure of G200, the first successful ionomer glass, was reported in 1979 (Barry et al. 1979). Although differing somewhat from modern ionomer glasses in that it is low in sodium and very high in fluoride, G200 was found to show some distinctive features that appear to be typical of glasses capable of forming satisfactory glass-ionomer cements. It was shown by scanning electron microscopy to contain phase-separated droplets of complex structure as well as substantial deposits of crystalline fluorite (Barry et al. 1979
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


