Systemic lupus erythematosus is a chronic autoimmune disorder characterized by production of autoantibodies directed against nuclear and cytoplasmic antigens, affecting several organs. Although cause is largely unknown, pathophysiology is attributed to several factors. Clinically, this disorder is characterized by periods of remission and relapse and may present with various constitutional and organ-specific symptoms. Diagnosis is achieved via clinical findings and laboratory examinations. Therapies are based on disease activity and severity. General treatment considerations include sun protection, diet and nutrition, smoking cessation, exercise, and appropriate immunization, whereas organ-specific treatments include use of steroidal and nonsteroidal anti-inflammatory drugs, immunosuppressive agents, and biologic agents.
Key points
- •
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder, characterized by unknown etiology and a multifactorial pathophysiology.
- •
Clinical manifestations are very heterogeneous, and may virtually affect any organ. The diagnosis of SLE is mainly based on clinical and Immunologic criteria.
- •
The management of SLE is complex and require a multidisciplinary approach. SLE pharmacological treatment can envisage the use of both biologic and non-biologic agents.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder, characterized by production of autoantibodies directed against nuclear and cytoplasmic antigens, which may affect several different organs, with a plethora of different clinical and immunologic abnormalities, characterized by a relapsing and remitting clinical course.
The history of lupus dates back at least to the thirteenth century, when the physician Rogerius described for the first time erosive facial lesions, mimicking a bite from a wolf ( lupus in ancient Latin means wolf). From the Middle Ages to the mid to late nineteenth century, the main clinical descriptions of lupus were dermatologic, as described by Bateman, Cazenave, and Kaposi. In 1833, Cazenave coined the term, erythema centrifugum , to describe cutaneous lesions that are now referred to as discoid lupus, while von Hebra in 1846 described the butterfly distribution of the facial rash. In 1872, Kaposi described for the first time the systemic manifestations of lupus, including subcutaneous nodules, arthritis with synovial hypertrophy of both small and large joints, lymphadenopathy, fever, weight loss, anemia, and central nervous system involvement, confirmed by Osler and Jadassohn several years later.
It is only in the mid-twentieth century, however, that remarkable scientific advances have been achieved, due to the discovery of the lupus erythematosus (LE) cell in the bone marrow of patients with acute disseminated LE in 1948 as well as the false-positive test for syphilis and the immunofluorescent test for antinuclear antibodies (ANAs).
During the following years, new autoantibodies, such as antibodies to DNA (anti-DNA), antibodies to extractable nuclear antigens (nuclear ribonucleoprotein [RNP], Sm, Ro, and La), and anticardiolipin antibodies, were recognized as causative of clinical subsets of lupus and helped better clarify the underlying pathogenesis.
Similarly, the development of animal models, the recognition of the role of genetic predisposition in some lupus families, and the ever-increasing number of targeted medications now represent important milestones in the overall understanding of this disorder.
Despite these new advances, SLE remains a clinical enigma for both patients and physicians due to its unpredictable course. In many cases, this disease may be mild and patients may live a normal life, whereas in many others, it is a devastating disease process. For this reason, a multidisciplinary management approach is often necessary for SLE patients.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder, characterized by production of autoantibodies directed against nuclear and cytoplasmic antigens, which may affect several different organs, with a plethora of different clinical and immunologic abnormalities, characterized by a relapsing and remitting clinical course.
The history of lupus dates back at least to the thirteenth century, when the physician Rogerius described for the first time erosive facial lesions, mimicking a bite from a wolf ( lupus in ancient Latin means wolf). From the Middle Ages to the mid to late nineteenth century, the main clinical descriptions of lupus were dermatologic, as described by Bateman, Cazenave, and Kaposi. In 1833, Cazenave coined the term, erythema centrifugum , to describe cutaneous lesions that are now referred to as discoid lupus, while von Hebra in 1846 described the butterfly distribution of the facial rash. In 1872, Kaposi described for the first time the systemic manifestations of lupus, including subcutaneous nodules, arthritis with synovial hypertrophy of both small and large joints, lymphadenopathy, fever, weight loss, anemia, and central nervous system involvement, confirmed by Osler and Jadassohn several years later.
It is only in the mid-twentieth century, however, that remarkable scientific advances have been achieved, due to the discovery of the lupus erythematosus (LE) cell in the bone marrow of patients with acute disseminated LE in 1948 as well as the false-positive test for syphilis and the immunofluorescent test for antinuclear antibodies (ANAs).
During the following years, new autoantibodies, such as antibodies to DNA (anti-DNA), antibodies to extractable nuclear antigens (nuclear ribonucleoprotein [RNP], Sm, Ro, and La), and anticardiolipin antibodies, were recognized as causative of clinical subsets of lupus and helped better clarify the underlying pathogenesis.
Similarly, the development of animal models, the recognition of the role of genetic predisposition in some lupus families, and the ever-increasing number of targeted medications now represent important milestones in the overall understanding of this disorder.
Despite these new advances, SLE remains a clinical enigma for both patients and physicians due to its unpredictable course. In many cases, this disease may be mild and patients may live a normal life, whereas in many others, it is a devastating disease process. For this reason, a multidisciplinary management approach is often necessary for SLE patients.
Epidemiology
Incidence and Prevalence
SLE usually affects adult women, although in 15% to 20% of cases, SLE may occur in childhood. In addition, childhood-onset SLE seems more severe than the adult-onset SLE, because it seems to have a more severe clinical course.
The actual incidence, prevalence, and gender distribution of SLE is still unknown due to the lack of a universal methodology adopted by all available studies, leading to interpreting such studies with caution. SLE seems to affect people of all races, genders, and ages, with a higher peak of incidence/prevalence among African Americans and African Caribbeans, with a predilection for women in their third to fourth decades of life.
Overall incidence is wide ranging, from 1.0 (per 100,000) in Denmark to 8.7 in Brazil, whereas the overall prevalence was estimated to be wide ranging, from 28.3 in Denmark to 149.5 in Pennsylvania. If these data are considered based on the ethnicity, however, the highest incidence and prevalence were reached by the African Caribbean population, as reported in 2 different UK studies. In the first, the incidence was 11.9 and the prevalence was 111.8, whereas in the second, the incidence was 31.9 and the prevalence was 207.0. Similarly, even in the United States, the incidence and prevalence in African Americans were 2-fold to 3-fold higher than in European Americans. Similarly, SLE is more common in Native Americans in Canada, in Maori and Pacific people in New Zealand, and in aborigines in Australia compared with the respective European populations (and their descendants) who live in the same regions. These findings show that Europeans and their descendants in different countries of the world have a lower incidence/prevalence of SLE.
Few data from Asia are available: the prevalence from 24 different Asian countries usually ranges from 30 to 50 per 100,000 population, whereas incidence rates, as reported from only 3 countries, varied from 0.9 to 3.1 per 100,000.
From Western Africa (the location of descendants of many African Americans) a low incidence/prevalence of SLE was reported, probably due to the presence of malaria, which could have altered the immune response, providing some sort of protection from autoimmunity. Because of the few data, solely based on sporadic case reports and case series, it is hard to claim the real epidemiology of SLE in that region.
As far as gender/age distribution is concerned, data are concordant, with women more frequently affected than men; however, conflicting data are present as far as age onset. For instance, the peak incidence among women in the United Kingdom was reported as 50 to 54 years, whereas in Quebec, Canada, it was 45 to 64 years. This incidence dramatically decreases in other countries of the world, ranging from 32.2 years in aborigines to 32.3 years in Hong Kong Chinese to 24 to 26 years in Malaysia to 25 years in Philippines to 20 years in Oman and to 25 years in Saudi Arabia.
Mortality Rate
Data from the most recent studies have shown that the survival rate of patients with SLE has dramatically increased over the past 50 years, from less than 50% in the 1950s to almost 95% at the beginning of the twenty-first century.
The mortality rate in SLE patients compared with healthy individuals, however, remains 2 to 4 times higher; in the United States, the survival rate ranges from 95% at 5 years to 78% at 20 years, with similar reported data from other countries, such as Latin America, Greece, and Saudi Arabia.
Lower survival rates after 5 years was seen in Tunisia, with 86%, in Pakistan, with 80%, and in South Africa, ranging from 57% to 72%.
Finally, several studies also have confirmed that the risk of mortality in African Americans was 2 to 3 times higher compared with European Americans and the mortality rate in Native North Americans was approximately 4 times higher than in non–Native American population.
Etiopathogenesis
As in many other autoimmune diseases, either the cause or the underlying pathophysiologic mechanism that triggers the autoimmune response in SLE remains largely unknown. Recently, several investigations have tried to hypothesize SLE etiology and clarify all possible genetic susceptibility and immunologic mechanisms involved in the pathophysiology of SLE.
Over the past 20 years, several investigations have demonstrated that a genetic susceptibility to SLE exists, showing a high concordance rate in monozygotic twins (24%–58%) compared with 2% to 5% in dizygotic twins. In addition, some studies showed that the frequency of SLE in relatives of SLE patients ranged from 8% to 10%, being similar among many populations, such as Europeans, Latin Americans, and African Americans, and being higher in Israeli Arab (24%) and Omani patients (48%).
Many candidate genes have been associated, however, with a high susceptibility of SLE. Genome-wide association studies have revealed approximately 30 to 40 gene loci with polymorphisms that predispose to SLE.
The strongest risk in all genome-wide association studies was related to the major histocompatibility complex, which contains genes for antigen-presenting molecules (human leukocyte antigens class I [A, B, and C] and class II [DR, DQ, and DP]).
In SLE patients, an increased susceptibility was found in
- •
HLA-DRB1, HLA-DQA1 in Europeans and Chinese
- •
HLA-DBQ1 in Europeans
- •
HLA-DR3 or DRB1*0301 in Europeans and their descendants in North America and Australia, Tunisians, and Eastern Indians
- •
HLA-DR4 in Northern Indians
- •
HLA-DR3 in Latin Americans
- •
HLA-DR8 in Europeans and in Hispanic Americans mostly from Mexico
- •
HLA-DR2 subtype DRB1*1501 in Europeans, Latin Americans, Tunisians, Japanese, Chinese, and Koreans
- •
HLA-DR2 subtype DRB1*1503 in African descendants
Conversely, 2 haplotypes (HLA-DR8 subtype DRB1*0802 and HLA-DRB5) were shown protective in Mexican and European/Latin Americans, respectively.
The role of HLA polymorphisms in SLE patients can be better understood in the light of pathogenic production of autoantibodies; some studies have shown that HLA alleles are not related to the SLE pathogenesis per se but to the production of specific autoantibodies and, in turn, to specific clinical manifestations. For instance, it seems that HLA-DR3 seems related to anti-Ro/SSA and anti-La/SSB production in European population, with subsequent renal and pulmonary involvement.
The role of HLA in SLE susceptibility seems, however, to be not the only genetic factor; different genes might be involved, such as IRF5, BLK, and STAT4 in Europeans and Chinese people, which seem to play an important role in the expression of interferon (IFN)-inducible gene. Approximately 60% of SLE patients were found to have an overexpression of IFN-α–induced genes. Also, some single-nucleotide polymorphisms in the IRF5 gene have been identified as risk factors in many populations (Europeans, European Americans, African Americans, Mexican, and Japanese). Some other single-nucleotide polymorphisms seem to predispose to particular clinical subsets of SLE; for instance, a single-nucleotide polymorphism in the STAT4 gene seems to predispose to high titer of anti-DNA autoantibodies, nephritis, and antiphospholipid syndrome.
Still other genes seem to play a role, such as variation in the interleukin (IL)-10 promoter or variation in gene copy number of complement component C4.
It seems that no single gene polymorphism is involved in the SLE pathogenesis; it is a combination of susceptibility genes and absence of protective genes. Even epigenetic factors, such as hypomethylation of DNA, may contribute to the SLE pathogenesis.
Possible Etiology
Recent investigations have hypothesized that a possible etiologic explanation of SLE might reside in the self-organized criticality theory. This theory states that an overstimulation of a host’s immune system by repeated exposure to several antigens that exceeds the immune system’s self-organized criticality may induce an autoimmune response.
Such antigens would be normally presented to T cells and then stimulate host CD4 + T cells to produce autoantibody-inducing CD4 + T cells, which in turn not only would stimulate B cells to produce a wide variety of autoantibodies but also foster the differentiation of CD8 + T cells into cytotoxic T lymphocytes via antigen cross-presentation. It seems that the capability of an SLE individual of developing an autoimmune response is determined (probably genetically) by ability of presenting and/or cross-presenting antigen to T cells efficiently.
Pathophysiology: A Paradigm of Autoimmunity
The general pathophysiologic mechanism in SLE is complex; tissue injury is promoted by several factors: both genetic and epigenetic factors (discussed previously), the production of autoreactive dendritic cells (DCs) (innate immunity), and the stimulation of autoreactive CD4 + T cells and B cells (adaptive immunity) in producing autoantibodies along with the employment of inflammatory cytokines (in particular, IFN) and hormonal and environmental factors.
Innate immunity
The role of innate immunity might be linked to a dysregulation of 2 different types of cells, DCs and neutrophils (NETs).
Although the precise mechanism needs to be better elucidated, it seems that DCs play a crucial role in expansion of both autoreactive T cells and B cells with autoantibody production in a murine model of lupus, indicating their role in promoting extrafollicular humoral response in SLE patients.
Dysregulation of NETs is another important key point in the SLE pathogenesis, because it seems that activated NETs in SLE patients die in a unique process called NETosis (apoptosis of NETs), making free a large amount of DNA in the form of weblike structures, so as to be protected by endonuclease and, thus, constituting an important source of autoantigens in SLE with uptake for DCs activation and subsequent pathogenic production of type I IFN.
Adaptive immunity
The function of autoreactive B cells and T cells in SLE pathogenesis is better understood. Some studies have proved that B-cell tolerance is aberrant and, in combination with enhanced B-cell receptor, Toll-like receptor, with B-cell activating factor receptor, may promote activation and survival of autoreactive B cells. Similarly, T cells (CD4 + ) play a crucial role in regulating B-cell response and infiltrating the target tissue, with subsequent damage.
Therefore, both types of immune cells cooperate to produce pathogenic autoantibodies. The role of autoreactive B cells in SLE pathogenesis is well understood, via the differentiation into pathogenic memory and plasma cells and subsequent production of autoantibodies, but the role of T cells is still under investigation. Recent studies in a mouse model and in humans have pointed out that follicular helper T cells are dysregulated, fostering the differentiation of B cells. In patients with active SLE nephritis, treatment with antibodies against CD154 inhibits disease activity and abnormal B-cell differentiation. Therefore, circulating immunoglobulin (Ig)-secreting cells (B cells) and, subsequently, serum anti–double-stranded DNA (anti-dsDNA) antibodies levels dramatically decrease.
Role of autoantibodies
The dysregulation of both innate and adaptive immunity promote the production of autoantibodies against approximately 150 different types of antigens, which are the initiations of tissue injury. The most implicated autoantibodies in the pathogenesis of SLE are those ones directed against the nuclear component (ANA) of cells, which include autoantibodies against DNA (single stranded and double stranded), chromatic, histone, Sm, SSA/Ro, SSB/La, and RNP as well as anti-C1q, phospholipid-associated proteins, cytoplasmatic molecules, endothelial-membrane antigens, complement fragments, and IFNs.
Intriguingly, among all different types of ANA autoantibodies, just anti-dsDNA was significantly associated with disease activity and specific organ damage in SLE. As in many other autoimmune diseases, as in SLE, both pathogenic or nonpathogenic autoantibodies coexist. Therefore, SLE patients have high titer of ANA, but people with a positive ANA titer do not necessarily have SLE.
The underlying pathogenic mechanisms by which autoantibodies exert their action is also complex and variable, including direct cellular lysis, cellular opsonization, immune complexes deposition, complement fixation, and progressive inflammation.
So far, it is hard to establish a conclusive association between specific autoantibodies and a clinical phenotype in SLE; currently, the most well-known association stands between lupus nephritis and anti-dsDNA antibodies and more recently between lupus nephritis and anti-C1q antibodies.
Recent studies have also demonstrated high titers of IgE in SLE patients without associate allergy, which, at least in a lupus-prone mice model, have provoked an immunocomplex-mediated nephritis.
Hormonal factors
Gender is one of the strongest risk factors in SLE onset: the female-to-male ratio is 9:1, with a peak in women during their reproductive life. This may reflect gonadal sex steroid alterations in the development of SLE in women. Several studies, mostly on sexual hormones in SLE patients, have demonstrated that hormonal imbalance may modulate the incidence and severity of SLE. Hormonal alterations were found in
- 1.
Sexual hormones: although they remain within the normal range, statistical differences have been found in SLE patients versus healthy controls. In women, dehydroepiandrosterone (DHEA), testosterone, and progesterone were found significantly lower in SLE patients compared with controls, whereas estradiol and prolactin were higher. Conversely, in men, DHEA may be low, progesterone unknown, and testosterone and estradiol normal, whereas prolactin was high. Further studies have also highlighted, for instance, that in breastfeeding women the incidence of SLE decreased, whereas nulliparous women seemed to run a higher risk of developing SLE.
- 2.
Thyroid hormones: elevated levels of antithyroid antibodies and serum THS level were detected in SLE patients.
- 3.
Hypothalamic axes: SLE patients seem to have an altered autonomic nervous system response, more pronounced during the human corticotropin-releasing hormone stress test.
Environmental factors
Several environmental factors have been considered as playing a role in SLE:
- 1.
Virus: Epstein-Barr is found in high titer in SLE children.
- 2.
Ultraviolet light can foster the expression of more small nuclear RNPs on cell surface and decrease the T-cell DNA methylation activity, with subsequent adaptive immunity imbalance and autoantibody formation.
- 3.
Silica dust and smoking cigarettes: both increase the risk of developing SLE, especially in African American women.
- 4.
Medications: several medications, such as penicillin, sulfa drugs, and codeine, have been reported as causative agents of SLE.
- 5.
Alcohol use: a moderate consumption seems to be a protective factor.
Clinical manifestations
SLE is an unpredictable disease, from a clinical standpoint: it is characterized by alternating periods of remission and exacerbation and by a wide variety of clinical manifestations (signs and symptoms) that may affect any organ.
SLE signs/symptoms may be divided in 2 major categories:
- 1.
Constitutional signs/symptoms
- 2.
Organ signs/symptoms
Constitutional Signs/Symptoms
- 1.
Fatigue is one of the most common symptoms and can be seen in almost all patients (80%–100%). It seems to not be correlated with disease activity per se but rather to other factors, such as depression, stress, anemia, smoking habit, sedentary living, sleep disturbances, and coexistent fibromyalgia.
- 2.
Fever can be found in more than 50% of SLE patients; it can be a manifestation of SLE per se or a symptom related to an SLE-induced infection or an adverse SLE drug reaction.
- 3.
Weight changes: weight loss is usually seen before the diagnosis of SLE, whereas weight gain seems to be related to corticosteroids—side effects are increased appetite and nephritic SLE electrolyte and albumin imbalance–related fluid levels.
Organ Signs/Symptoms
- 1.
Musculoskeletal signs/symptoms usually include arthralgia, arthritis, osteonecrosis avascular necrosis of bone, and myopathy. The arthritis and arthralgias of SLE tend to be migratory, usually involving symmetrically small joints, such as knees, carpal joints, and fingers, especially the proximal interphalangeal joint. All joints, however, may be affected by SLE. Also, periarticular structures can be inflamed, leading to tendonitis, tenosynovitis, and tendon rupture. Avascular necrosis can also occur, causing joint pain and disability, mostly in larger joints, such as hip and knee. Lastly, muscles are often affected—myalgia and muscle weakness, mostly in the neck, pelvis, thighs, shoulders, and upper arms, makes it difficult for patients to climb stairs and/or get up from a chair, for instance.
- 2.
Renal signs/symptoms can be due to a direct damage from SLE to kidney or a kidney damage due to drug toxicity. Lupus nephritis remains one of the most important SLE manifestations, causing substantial morbidity and mortality. The most common form of lupus nephritis is represented by glomerulonephritis from pure mesangial alteration to end-stage fibrosis, frequently accompanied by tubulointerstitial and/or vascular lesions. A wide variety of other lesions may be present, such as renal amyloidosis, focal segmental glomerulosclerosis, IgA and IgM nephropathy, and necrotizing glomerulitis.
- 3.
Gastrointestinal signs/symptoms are common in SLE patients; one of the most common causes of gastrointestinal complaints is mesenteric vasculitis and thrombosis, which can lead to life-threatening ischemia, perforation, and infarction. Other gastrointestinal issues in SLE patients are protein-losing enteropathy, intestinal pseudo-obstruction, acute pancreatitis, and, more rarely, celiac disease, inflammatory bowel diseases, eosinophilic enteritis, and pneumatosis cystoides intestinalis.
- 4.
Pulmonary: lungs in SLE patient are usually affected later in the course of the disease along with other organs. The most common pulmonary manifestation attributable to SLE is pleuritis ; however, even parenchymal disease, such as pneumonitis, acute respiratory distress, diffuse alveolar hemorrhage, chronic interstitial pneumonitis, and shrinking, or vanishing, lung syndrome can be seen. Similarly seen, although more rarely, is vascular involvement: acute reversible hypoxemia, pulmonary embolisms, pulmonary arterial hypertension, and airway disease obstructive lung disease and upper airways disease.
- 5.
Cardiovascular: several studies have already demonstrated a clear association between SLE and cardiovascular disease (CVD). The main CVDs of SLE are valvular heart diseases associated with Libman-Sacks disease lesions, sterile vegetations, serositis associated with pericardial disease, and venous and arterial thrombosis associated with antiphospholipid antibodies. It seems that such association between SLE and CVD strictly depends on a combination of several risk factors for CVD, including dyslipidemia and, to a varying degree, hypertension, diabetes, smoking, inflammation, lipid oxidation, antiphospholipid antibodies, and renal disease and renal failure.
- 6.
Neuropsychiatric: the central nervous system is often affected in SLE patients, with the development of both neurologic and psychiatric symptoms. The 2 most common neurologic manifestations in SLE patients are seizures and cerebrovascular disease, such as stroke, transient ischemic attack, and venous sinus thrombosis, whereas the 2 most common psychiatric manifestations are depression and cognitive dysfunction. Many other additional symptoms, however, can be present in SLE patients, such as headaches, mood disorders, acute confusional states, and anxiety. A recent study has shown that SLE patients may develop the following events: seizure disorder (28%), cerebrovascular disease (19%), acute confusional state (14%), psychosis (11%), myelopathy (8%), mood disorder (6%), headache (4%), movement disorder (2%), cranial neuropathy (3%), demyelinating syndrome (1.5%), anxiety disorder (1.5%), mononeuritis multiplex/mononeuropathy (1.5%), aseptic meningitis (1%), and polyneuropathy (1%).
- 7.
Hematological: blood disorders are common in SLE patients and can present with significant clinical manifestations. The principal hematological issues are cytopenia and thrombophilia. SLE patients may have abnormalities, including anemia, thrombocytopenia, neutropenia, and leukopenia. In addition, they have an increased risk of thromboembolic events, particularly SLE patients with antiphospholipid antibodies—anticardiolipin, anti-β 2 -glycoprotein I antibodies, and lupus anticoagulant (LA).
- 8.
Ocular: approximately 30% of SLE patients suffer from ocular involvement, which includes involvement of the optic nerve, periorbita, and ocular adnex, such as orbit, conjunctiva, and eyelids. The most common manifestation is keratoconjunctivitis sicca, in which one or both eyes may have persistent dryness. The most devastating symptoms are secondary to optical nerve damage optic neuritis and ischemic optic neuropathy and retinal vaso-occlusion that is similar to diabetic and hypertensive retinopathy, which then may seriously threaten visual acuity.
- 9.
Cutaneous: the skin is one of the most affected organ in SLE patients (85%) and can be the only organ involved, as in cutaneous LE. Clinically, the mucocutaneous manifestation in SLE patients may be divided in 3 major groups: (1) chronic cutaneous LE (CCLE), (2) subacute cutaneous LE (SCLE), and (3) acute cutaneous LE (ACLE). The associated disease severity is usually evaluated by the cutaneous LE disease area and severity index. Diagnosis of mucocutaneous SLE needs to be made via histopathologic and immunohistological examinations. Cutaneous manifestations include a wide broad spectrum of different clinical features, which are considered important for diagnosis (discussed later). The main cutaneous manifestations are as follows :
- •
Malar rash consists of an erythematous rash over the cheeks and nasal bridge, with a butterfly shape and which sometimes can be painful or pruritic ( Fig. 1 )
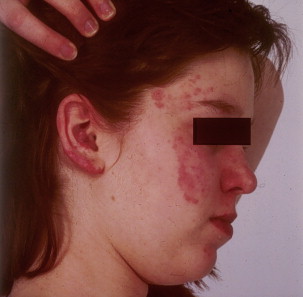
Fig. 1 Erythematous malar rash of a patient with systemic lupus erythematosus. Erythematous lesion is also present on patient’s ear. - •
Discoid lesions (localized and generalized) are disc-shaped lesions with erythematous plaques of different size with follicular hyperkeratoses. These lesions spread centrifugally, may merge, and are sometimes painful and pruritic. Some lesions may become hypertrophic. Rarely they may transform into skin cancer. These lesions represent the most common form of cutaneous LE, called discoid LE.
- •
Photosensitivity is the possibility of diffuse erythematous rash in sun-exposed areas, such as faces, arms, and dorsal part of the hands, sparing the knuckles
- •
SCLE: this form of LE is characterized by papular/plaque lesion with a slight scaling, which can be confluent, forming papulosquamous or annular and/or polycyclic lesions. Such lesions are usually asymptomatic and can be exacerbated by sun exposure. This subset of SLE is usually benign and carries a good prognosis.
- •
Alopecia is detectable in approximately 45% of patients and usually affects the temporal regions, causing a patchy pattern of hair loss. It may be one of the first symptoms of SLE.
- •
Lupus panniculitis/lupus profundus: this form of SLE is rare (approximately 2%) and it consists of deep brawny indurations or subcutaneous nodules, in which the epidermis is usually erythematosus, atrophic, or ulcerated. These lesions normally affect proximal extremities, in particular arms and shoulders.
- •
Atrophie blanche is a particular type of scar arising on the lower legs, which occurs when skin is injured and the blood supply is inadequate. It is usually characterized by painful, petechial, pruritic papules or hemorrhagic bullae.
- •
Livedo reticularis consists of lacelike purplish violaceous discoloration frequently seen on the lower extremities, probably caused by swelling of medium veins in the skin, and can be aggravated by cold exposure.
- •
Other cutaneous manifestations include periungual telangiectasias, lichen planus in LE, small vessel cutaneous leukocytoclastic vasculitis, Raynaud syndrome, and bullous lesions.
Oropharyngeal Manifestations
Oropharyngeal manifestations are one of the most important clinical aspects of SLE patients, because they represent one of the criteria for diagnosis. Oral lesions in SLE, however, are not always a constant finding, with a frequency ranging from 9% to 45% in systemic disease and from 3% to 20% in localized cutaneous disease ( Fig. 2 ).
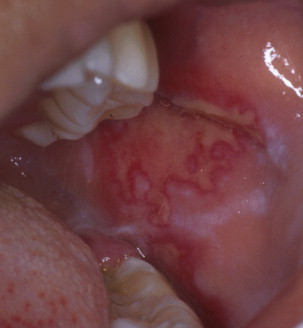
Oral lesions may be seen in all 3 forms of cutaneous LE :
- •
CCLE: oral lesions are usually represented by well-demarcated, round, or irregular red lesions that can be atrophic or ulcerated with a white radiating keratotic striae and telangiectasias. They may also assume, however, a verrucous aspect or be linear fissured or have ulcerative lesions. Such lesions, unlike lichen planus lesions, are asymmetrically distributed and the hard palate seems the most affected oral site, followed by the lips, in which lesions tend to go beyond the vermillion border and affect the surrounding skin. Rarely, squamous cell carcinoma may occur on longstanding scarring lesions.
- •
SCLE: oral lesions in this form are rare, because SCLE is highly associated with photosensitivity. They are characterized by red, round, discretely atrophic patches mainly located on the hard palate. Less frequently, lips are affected by diffuse erythematous scaling plaques, which tend to invade the surrounding skin.
- •
ACLE: oral lesions in this form are usually represented by ulcerations, mainly on the hard palate accompanied by an intense erythema and petechiae. In addition, erythemato-purpuric macules can been seen mostly on labial mucosa, which look like those present on the skin but may appear even in the absence of skin lesions. Lastly, the presence of multiple blisters affecting virtually the entire oropharyngeal cavity may be seen in the bullous form of SLE.
Other oropharyngeal signs and symptoms possibly present in SLE patients include oral candidiasis, with a prevalence ranging from 4% to 75%; dysphagia, with a prevalence ranging from 11% to 75%; and xerostomia, with a prevalence from 1% to 100%. This last symptom, however, might not be simply an oral SLE symptom but may be due to secondary Sjögren syndrome.
A recent meta-analysis has highlighted that the prevalence of Sjögren syndrome in SLE patients is estimated to be approximately 17.8%, and SS could be diagnosed in a period ranging from 0.33 to 10.8 years after the diagnosis of SLE. Patients with SLE–Sjögren syndrome seem to have less internal organ involvement and have a more favorable overall prognosis. In half of the studies considered in this review, however, the percentage of patients with oral involvement (mainly oral ulcer) was higher in SLE–Sjögren syndrome that in SLE only. In cases of suspected oral SLE, a differential diagnosis formulation with many other diseases is mandatory and includes oral lichen planus and lichenoid lesions, erythema multiforme, pemphigus vulgaris, mucous membrane pemphigoid, herpes simplex, fungal infections, and syphilis.
Histopathology examination on oral biopsy shows alteration in the epithelium, basement membrane zone, and the lamina propria. The epithelium usually presents with hyperorthokeratosis or hyperparakeratosis, granulosis, acanthosis, spongiosis, hydropic degeneration with presence of colloid bodies, and hyperproliferation of basal keratinocytes. The basement membrane zone shows focal thickening, whereas the lamina propria contains a predominant mononuclear lymphocytic infiltrate with focal or diffuse aggression to the basal keranotinocyes. Such inflammatory infiltrate usually affect the superficial lamina propria but may also cause vasculitis in the deeper lamina propria, with thickening of blood vessel walls and the presence of a severe perivascular inflammatory cell infiltrate. Lastly, the presence of mucin in the lamina propria is considered an important marker distinguishing this disease with lichen planus.
Direct immunofluorescence analysis shows a deposit of IgM and complement fraction C3 along the basement membrane zone in linear or granular pattern as well as apoptotic (colloid) bodies.
Diagnosis
The diagnosis of SLE is mainly based on 2 main criteria:
- •
Clinical
- •
Immunologic
Sometimes, an immunohistopathologic examination is needed in cases of a specific organ involvement (eg, skin or kidney). The results from hematoxylin-eosin stain and direct immunofluorescence can give a strong suggestion of SLE. A careful differential diagnosis, however, among many other systemic diseases is warranted.
Both clinical and immunologic criteria are considered mandatory in order to establish a definitive diagnosis of SLE, according to the American College of Rheumatology (ACR), which revised the diagnostic criteria for SLE, proposed in 1982, and to the Systemic Lupus International Collaborating Clinics (SLICC) group. The differences between the 2 classification criteria are summarized in Table 1 .


