Hematopoietic cell transplantation is used to treat malignancies, hematologic and immune deficiency states, marrow failure syndromes, and autoimmune diseases. Graft-versus-host disease (GVHD) is a clinical syndrome seen following allogeneic transplantation where donorderived immunocompetent T cells and inflammatory responses attack host tissues. GVHD can cause significant morbidity and even result in mortality. The oral cavity is a frequently involved site with clinical changes resembling autoimmune collagen vascular diseases. Recognition, diagnosis, and monitoring of oral GVHD can help with diagnosis and grading of GVHD and judging responses to therapy. Topical and local management of symptomatic oral GVHD can reduce oral symptoms that can interfere with oral function and quality of life, and can reduce the need for more intensive immunosuppressive systemic therapies.
Hematopoietic cell transplantation (HCT) began in the late 1950s and has seen a steady increase in the overall success and applicability to treat a wide range of malignancies, hematologic and immune deficiency states, and autoimmune diseases . Animal studies in the late 1940s and early 1950s laid the ground work for HCT, and human HCT was first attempted in the late 1950s. However, early HCT attempts were complicated by significant and often lethal complications including infection, graft failure, relapse, hemorrhage, and, in the case of allogeneic HCT (alloHCT), graft-versus-host disease (GVHD) . GVHD is a clinical syndrome where donor-derived immunocompetent T cells react against patient tissues directly or through exaggerated inflammatory responses following alloHCT . The primary target organs of GVHD classically have been those of skin, liver, and the gastrointestinal (GI) tract. However, the oral cavity is also frequently involved, possibly only second to cutaneous involvement . Despite significant advances, GVHD remains a major cause of morbidity and mortality with chronic GVHD being the leading cause of nonmalignant fatality post alloHCT .
Given the impact of GVHD on the allogeneic patient’s post HCT course, timely and accurate diagnosis of oral GVHD, ongoing assessment of responses to therapy, and the appropriate management of oral GVHD can contribute to not only improved patient comfort, oral health, and function, but possibly long-term survival . Since the oral cavity is frequently involved and is easily assessed for GVHD, recognition of oral GVHD changes can contribute to improved post-HCT medical management. Finally, appropriate therapy for symptomatic oral GVHD can significantly improve the patient’s quality of life and overall oral function.
Pathobiology, epidemiology, and clinical manifestations of graft-versus-host disease
In 1966 Billingham formulated the three fundamental elements required for the occurrence of GVHD. First, the transplanted graft must contain immunologically competent cells; second, the recipient must be incapable of rejecting the transplanted cells; and third, the recipient must express tissue antigens that are not present in the donor . In the most simplistic terms, based on these fundamental principles, GVHD is a clinicopathologic syndrome that occurs following alloHCT when transplanted immunologically competent donor (graft) T cells recognize and react against histocompatability antigens on the patient’s cells (host) and induce immune responses resulting in host tissue damage . These processes are triggered by the recognition of host human leukocyte antigens (HLAs) by donor lymphocytes as being “foreign” antigens; the ensuing complex set of both autoimmune and alloimmune responses and severe inflammatory manifestations are recognized clinically as GVHD .
The most important risk factor for GVHD is the degree of HLA match of donor to patient. Mismatching in HLA-A, -B, -C, or -DRB1 will increase the risk of GVHD . Sibling HLA-matched (ie, related) donor grafts have less risk for GVHD than HLA-matched unrelated donor grafts, possibly because of mismatched minor histocompatibility antigens (mHA) that have been shown to influence GVHD incidence. Further immune-based differences that influence the risk for GVHD are being studied . Additional risk factors for GVHD include increasing patient age, donor parity and sex mismatch, choice of graft source (ie, peripheral blood stem cells, bone marrow, or umbilical cord), and pre-infusion graft modulations/manipulations, most notably T-cell depletion . Finally, the toxicity of high-intensity conditioning regimens, especially those that use total body irradiation, can increase GVHD risk because they cause more host tissue damage that results in enhanced recognition of host antigens by donor antigen-presenting cells (APCs) leading to increased activation of donor T cells.
GVHD was initially classified as either acute GVHD (aGVHD) or chronic GVHD (cGVHD) based on an arbitrary time point with aGVHD occurring within 100 days of transplantation and cGVHD occurring over 100 days after transplantation. Recently, there has been a shift toward separation of acute and chronic forms of GVHD based on clinical and pathological characteristics . While both forms represent the consequence of damage to host tissues by activated donor-derived T lymphocytes in response to the major histocompatibility complex (MHC) disparities between the donor and the host, elucidation of the immunopathobiology of GVHD has made it apparent that the specific pathophysiological mechanisms are distinctly different . Acute GVHD can occur as early as 1 week post-HCT or following donor lymphocyte infusion (DLI, see next paragraph). In contrast cGVHD can have an onset of 70 days or later post-HCT or DLI and continue for many years. There are four patterns of onset for cGVHD: (1) a de novo onset (without prior aGVHD), (2) quiescent onset (onset is after a period of no apparent aGVHD activity between the resolution of aGVHD and the onset of cGVHD), (3) progressive onset (cGVHD evolves directly from aGVHD), and (4) explosive onset (manifesting with an abrupt onset of severe multisystem involvement with manifestations of both acute and chronic GVHD) .
While both forms of GVHD contribute to significant post-HCT morbidity and mortality, for patients transplanted for malignancies, GVHD has also been associated with lower relapse rates because of a graft-versus-leukemia (GVL or graft-versus-tumor, GVT) effect . Chronic GVHD in particular has been shown to be associated with lower relapse rates, especially for patients who are transplanted in relapse . The GVL effect is thought to be mediated, at least in part, by donor T cells; however, the precise mechanisms that result in GVHD and GVL remain to be elucidated. Recognition of this beneficial effect has led to the use of a technique for patients who relapse post-HCT in which lymphocytes are collected from the donor and infused into the patient (referred to as a DLI). The goal of this treatment is to induce a T-cell–mediated GVL effect and has proven to be successful in many instances . Unfortunately, DLIs will also induce GVHD with its associated morbidity and mortality.
Acute graft-versus-host disease
Acute GVHD has an incidence rate of approximately 20% to 48% for matched related-donor HCTs, up to 70% for matched unrelated-donor HCTs, and MHC-mismatched (HLA haploidentical) patients have rates as high as 80% to 90% . If related donors are mismatched for 1-, 2-, or 3- HLA antigens, the risk of GVHD ranges from 75% to 80% . Acute GVHD generally occurs within 14 to 35 days of stem cell infusion but can be seen as early as within 1 week of transplant . As noted above, the incidence and severity of aGVHD is related to several immunologically based factors, such as donor-recipient HLA disparity, donor-recipient gender differences, source of stem cells, number of T cells in donor stem cell infusion, and effectiveness of GVHD prophylaxis regime . In patients receiving conventional GVHD prophylaxis, such as a combination of cyclosporine and methotrexate, the median onset of GVHD is 21 to 25 days after HCT, however onset may be delayed in T-cell depleted grafts . Reduced intensity conditioning regimens (previously referred to as nonmyeloablative conditioning regimes) may further decrease the incidence of aGVHD . A hyperacute form of GVHD may occur in patients with severe HLA mismatches and in patients who receive inadequate GVHD prophylaxis . Hyperacute GVHD is a severe form of aGVHD that can occur in the first 1 to 2 weeks after HCT and can be rapidly fatal .
The pathobiology of aGVHD has been described as a three-step process in which the innate and adaptive immune systems interact: (1) tissue damage to the recipient occurs from the radiation/chemotherapy conditioning regimen; (2) donor T cells recognize host antigens from the damaged tissues as foreign and become activated and stimulated, and then clonally expand; and (3) an effector stage ensues that is characterized by damage to host tissues induced either directly by immune cells or through a series of complex immune and inflammatory responses. In step one, conditioning regimens directly damage host tissues releasing alloantigens and inflammatory cytokines that promote activation of host APCs . In step two, host APCs present the host’s alloantigens to the resting donor T cells, thus activating them. Donor T-cell activation is characterized by cellular proliferation and the production of inflammatory cytokines, including interleukin (IL)-2, tumor necrosis factor alpha (TNF-α), and interferon gamma (INF-γ). Step three is characterized by multiple cytotoxic effectors including inflammatory cytokines, cytotoxic T cells, natural killer cells, other cells (mononuclear phagocytes and neutrophils), and nitric oxide production, producing direct target tissue damage or through intense inflammation associated with intense cytokine production, the so-called “GVHD cytokine storm” . The initial inflammation results in the further recruitment of effectors cells into target organs, amplifying local tissue injury with further secretion of inflammatory cytokines, which, together with cytotoxic T lymphocytes, lead to target tissue destruction . Additionally, specific proliferating marrow cells (eg, NK1.1 + T-cells and NK1.1 – T cells) are capable of suppressing or promoting T-cell responses and thus modulate the incidence and severity of aGVHD . Interestingly enough, donor-host histocompatibility differences may not always be needed to produce GVHD. A GVHD-like syndrome, seen rarely following autologous HCT, appears to arise through the inappropriate recognition of self-antigens . This is usually a mild self-limiting condition that readily responds to treatment with steroids.
The classically reported target organs for clinical manifestation of aGVHD are skin, liver, and the GI tract, but oral manifestations have clearly been documented . In the skin, a pruritic skin rash is noted with generalized erythoderma and in severe cases bullae formation and desquamation. Damage to the GI tract from conditioning regimen toxicity increases the translocation of inflammatory stimuli such as endotoxins, which promotes further inflammation and additional damage. Signs and symptoms of GI aGVHD include nausea, vomiting, diarrhea, and pain. Hepatic aGVHD is characterized as a cholestatic jaundice with a marked rise in bilirubin levels. Acute GVHD may also affect hematopoiesis resulting in a reduction in peripheral blood counts, particularly platelets .
Chronic graft-versus-host disease
cGVHD is the most common late complication after alloHCT and is the leading cause of late HCT non–relapse-related mortality . cGVHD prevalence varies from 25% to 80% in long-term survivors after alloHCT with 5-year survival rates as low as 40% for patients with severe multisystem cGVHD . The incidence and severity of cGVHD are correlated to immunologically related factors including HLA disparity, donor/host age and sex, donor type, source of progenitor cells, graft manipulations (especially T cell depletion), previous aGVHD, and use of post-HCT DLIs . Often, cGVHD will present during tapering of or soon after stopping aGVHD prophylaxis or treatment .
The pathophysiology of cGVHD is not well understood. There are two theories regarding the mechanism of cGVHD: the first is simply end-stage alloreactivity and the second is that cGVHD is caused by poor/dysfunctional immunologic recovery with the evolution of autoreactive T lymphocytes because of lack of thymic control . cGVHD is a multisystem alloimmune and autoimmune disorder characterized by immune dysregulation, immunodeficiency, and impaired organ function, all of which negatively impact survival . The pathobiology of cGVHD starts with the expansion of donor T cells in response to alloantigens or autoantigens that is unchecked by normal thymic or peripheral mechanisms of deletion. T cells promote target organ damage either directly through inflammatory cytokines, cytolytic attack and fibrosis, and/or by promoting B-cell activation and production of autoantibodies . While there can be widespread organ damage, the leading cause of death for patients with cGVHD is infection .
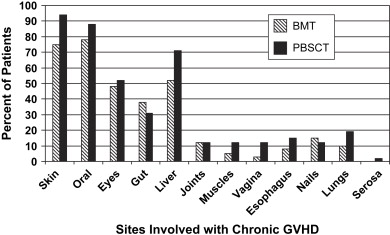
The most common sites of cGVHD involvement are the skin, oral cavity, eyes, GI tract, liver, and lungs ( Fig. 1 ); however, the spectrum of clinical involvement is remarkably variable . Clinical features are similar to those of collagen vascular diseases, lichen planus (LP), Sjögren syndrome, polyserositis, esophagitis and stricture, vaginal ulcerations and stricture, intrahepatic obstructive liver disease, obstructive pulmonary disease, progressive systemic sclerosis, fasciitis, and myositis .