Cranial sutures represent a form of bone articulation in which the margins of the cranial vault bones are connected by a thin layer of fibrous tissue. The cranial vault is composed of six major sutural areas ( Figure 44-1 ) and several minor sutures that serve two critical functions during the postnatal period. Initially the sutures allow head deformation during vaginal delivery as part of the birthing process. Later, during an infant’s postnatal development, cranial vault sutures facilitate head expansion to accommodate propulsive brain growth. Only small amounts of pressure (5 mm Hg) from the growing brain are required to stimulate bone deposition at the margins of a cranial bone. Under normal conditions, the brain volume will triple within the first year of life, and by age 2, the cranial capacity is four times that at birth.
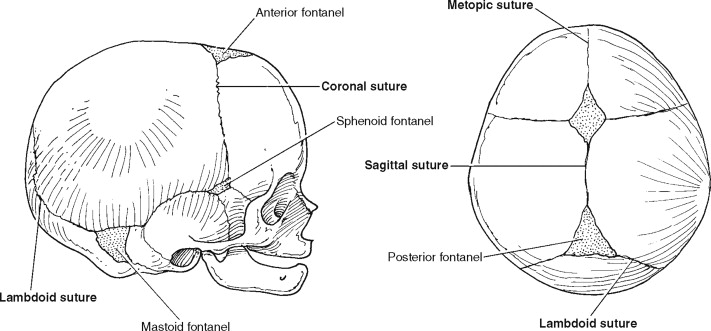
The term craniosynostosis is defined as a premature fusion of one or more of the cranial vault sutures. With rare exception, this is an intrauterine event so that a more accurate description of craniosynostosis may be a congenital absence of the cranial vault suture(s). The result is fusion of the bones adjacent to the suture and arrested sutural growth of the adjacent bones. The classic theory known as Virchow’s law states that premature fusion of a cranial vault suture results in limited development of the skull perpendicular to the fused suture and a compensatory “overgrowth” through the sutures that remain open. The result is a characteristic dysmorphology depending on which of the sutures is affected and potential neurologic consequences related to any underlying brain compression. Most forms of craniosynostosis represent random, nonsyndromic malformations limited to the cranial vault and orbital regions. Management typically requires a combined neurosurgical and craniofacial approach for release of the involved suture so that unrestricted brain growth and reshaping of the dysmorphic skeletal components can occur. In this chapter, the current diagnostic and surgical treatment approaches for patients with nonsyndromic craniosynostosis is presented. Perioperative considerations and specific surgical maneuvers used to treat the different forms of craniosynostosis are outlined with clinical examples.
FUNCTIONAL CONSEQUENCES OF CRANIOSYNOSTOSIS
Brain volume expansion occurs at a rapid rate during the first 2 years of life. During this time, it is growth of the visceral structures (i.e., brain and eyes) that drives the growth of the skeletal structures (i.e., cranial vault and orbits). Patent cranial vault sutures create a flexible complex of cranial bones that allow the growing brain to push them outward. Cranial growth occurs both at these open regions in a direction that is perpendicular to each suture and through endocranial resorption in combination with ectocranial deposition of bone.
INTRACRANIAL PRESSURE
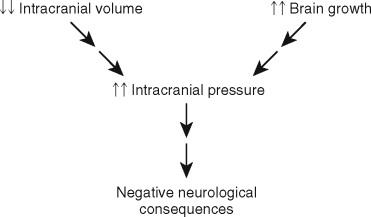
When a cranial vault suture is congenitally absent, the result is that the two adjacent cranial bone plates are fused together and growth along that suture is arrested. The combination of a rapidly expanding brain and diminished intracranial volume secondary to the restricted cranial growth may result in increased intracranial pressure (ICP) (greater than 15 mm Hg) with negative neurologic consequences. This sequence of events ( Figure 44-2 ) is traditionally used to explain the theoretic relationship between craniosynostosis and brain insult, but it does not adequately explain why some patients will develop elevated ICP and others will not. Previous investigations have determined that approximately 14% of children with untreated single-suture craniosynostosis will demonstrate increased ICP. When two or more sutures are fused, the likelihood of elevated ICP increases to 42%. This is the most concerning functional consequence of craniosynostosis since it may adversely affect brain function.
Children with untreated craniosynostosis who go on to develop increased ICP may exhibit a number of neurologic changes, including headaches, vomiting, sleep disturbances, feeding difficulties, behavioral changes, and diminished cognitive functioning. Symptoms related to increased ICP are rarely encountered before 1 year of age; frequently have a slow, gradual onset; are difficult to detect; and left untreated may be irreversible. Recognition of these neurologic signs and symptoms is further complicated by the young age of the patients with craniosynostosis who cannot yet communicate to alert parents and examiners of these physical findings.
HYDROCEPHALUS
Hydrocephalus is not usually observed in patients with nonsyndromic single-suture craniosynostosis, but it may occur independently and not necessarily as a consequence of this condition. In contrast, hydrocephalus is encountered in approximately 10% of children with multiple-suture craniosynostosis and may be seen in the craniofacial dysostosis syndromes. The correct diagnosis may require serial neurosurgical evaluations, computed tomography (CT) scan, magnetic resonance imaging (MRI), and cerebrospinal fluid (CSF) flow studies.
In cases where the infant has both hydrocephalus and craniosynostosis, careful consideration must be given to the exact timing and sequence of the cranial vault reshaping and ventriculoperitoneal (VP) shunt placement. Although the final decision is a neurosurgical one based on the child’s neurologic condition, placement of the VP shunt should be performed as a separate surgery after the cranial vault reconstruction when possible. The presence of a VP shunt at the time of synostosis repair may decrease brain expansion in the postoperative period thus reducing physical support to the bone segments and delaying the elimination of extradural dead space.
OPHTHALMOLOGIC EFFECTS
In patients with craniosynostosis and associated elevated ICP, the optic nerve is at risk. Prolonged, untreated ICP elevations will produce papilledema, optic nerve atrophy, and eventual loss of vision, which may be complete.
When the specific form of craniosynostosis produces orbital deformity or asymmetry, a negative impact on visual function may be realized. For example, orbital dystopia secondary to unilateral coronal synostosis can result in disturbances of extraocular muscle movement (i.e., strabismus), upper eyelid ptosis, and poor binocular vision. In cases of syndromic or nonsyndromic bilateral coronal suture craniosynostosis, decreased orbital volume will cause proptosis with an increased risk of direct trauma to the eye and corneal exposure or ulceration.
DIAGNOSTIC APPROACH TO ABNORMAL HEAD SHAPE
When an infant has a cranial vault asymmetry, the examiner must be alert to the possibility of prematurely fused sutures. However, abnormal head shape may be produced by various causes that must be distinguished from craniosynostosis. Most commonly, external forces applied to a flexible cranium with open sutures causes a “deformation” of the skull. In newborns, this deformational plagiocephaly is often the direct result of the head passing through the birth canal or the baby’s early decent into the true pelvis and may involve the anterior and posterior cranial vaults. Another common cause of deformational plagiocephaly are the postnatal external forces that result from repetitive sleep positioning. In 1992 the American Academy of Pediatrics made the recommendation that infants be placed on their backs during sleep to reduce the risk of sudden infant death syndrome (SIDS). Because of the “Back to Sleep” campaign and an increased awareness of cranial vault deformities within the pediatric community, an increased number of referrals for management of nonsynostotic posterior plagiocephaly has resulted. Accurately differentiating which cranial vault deformities are the result of nonsynostotic causes (i.e., positional plagiocephaly) and which are associated with a true absence of a cranial vault suture (i.e., craniosynostosis) is critical to establishing a diagnosis and formulating the appropriate treatment plan. The importance of a correct diagnosis is underscored by the dramatically different treatment approach to each condition. Craniosynostosis requires surgical treatment, whereas benign positional skull molding (positional plagiocephaly) is not associated with any neurologic consequences and typically requires conservative nonsurgical management using custom-made cranial orthotic devices or in the case of torticollis, neck physical therapy.
A careful history and clinical examination are the primary basis upon which the diagnosis of craniosynostosis is made. Because craniosynostosis is an intrauterine event, parents will often report that the deformity was noted immediately after birth and never improved after that time. By contrast, the typical history provided in cases of positional plagiocephaly is that the child’s head was well rounded at the time of birth and the asymmetry was noted 3 to 6 months later, suggesting postnatal deformation of a flexible cranial vault. Clinical examination of the infant with craniosynostosis is a key element in establishing the diagnosis. Each form of craniosynostosis will produce a unique dysmorphology that resulted from arrested skeletal growth perpendicular to the affected suture and continued or compensatory growth at the sutures that remain open. This combination of arrested growth in one region and additional growth in others may explain how unilateral sutural involvement results in a bilateral deformity and how fusion of one of the sutures within the posterior cranial vault will produce a deformity that affects the entire anteroposterior (AP) dimension of the cranium. An examiner familiar with these aberrant growth patterns and the characteristic dysmophologies specific to each type of synostosis will be able to consistently establish an accurate clinical diagnosis.
The diagnosis of craniosynostosis is made primarily on the basis of a careful clinical examination, but must be confirmed radiographically. Following clinical evaluation by a pediatric craniofacial surgeon and/or pediatric neurosurgeon, a complete skull series of plain radiographs is obtained to evaluate the cranial vault sutures. Standard views of the cranial vault are often all that is required to establish the absence of a cranial vault suture and confirm the diagnosis of craniosynostosis. In cases where all of the sutures cannot be adequately visualized using plain films, additional imaging with the use of CT is indicated. In addition to confirming the diagnosis, high quality craniofacial CT scans provide more detailed 3-D morphologic information, which is useful during the surgical planning phase. One-millimeter axial and coronal cuts with overlap and 3-D reconstructions of the cranial vault, cranial base, orbits, and maxillofacial skeleton are recommended. Precise patient positioning is important in obtaining a diagnostic scan, and this may require sedation or general anesthesia when the patient is an infant or young child.
Although technologic advancements have dramatically increased the accuracy of standard radiographic studies and CT imaging, incorrect and outdated terminology is still occasionally used in describing cranial vault sutures. At some medical centers, radiologists and surgeons continue to use terms such as “fibrous synostosis,” “suture dysfunction,” and “impending synostosis” when describing cranial vault sutures that otherwise appear open. We now know that craniosynostosis is in the vast majority of cases an intrauterine event that is either present at birth or not at all. Therefore if a suture appears patent on a radiographic study that is of diagnostic quality, the diagnosis of craniosynostosis is ruled out. Although the postnatal fusion of cranial vault sutures has been described in the scientific literature, it is considered an extremely rare event that is specifically related to specific clinical and syndromic conditions. The use of the terms described earlier is not consistent with contemporary knowledge about craniosynostosis, may be confusing to parents and health care providers, and may even result in inappropriate surgical treatment for patients who do not have craniosynostosis.
DIAGNOSTIC APPROACH TO ABNORMAL HEAD SHAPE
When an infant has a cranial vault asymmetry, the examiner must be alert to the possibility of prematurely fused sutures. However, abnormal head shape may be produced by various causes that must be distinguished from craniosynostosis. Most commonly, external forces applied to a flexible cranium with open sutures causes a “deformation” of the skull. In newborns, this deformational plagiocephaly is often the direct result of the head passing through the birth canal or the baby’s early decent into the true pelvis and may involve the anterior and posterior cranial vaults. Another common cause of deformational plagiocephaly are the postnatal external forces that result from repetitive sleep positioning. In 1992 the American Academy of Pediatrics made the recommendation that infants be placed on their backs during sleep to reduce the risk of sudden infant death syndrome (SIDS). Because of the “Back to Sleep” campaign and an increased awareness of cranial vault deformities within the pediatric community, an increased number of referrals for management of nonsynostotic posterior plagiocephaly has resulted. Accurately differentiating which cranial vault deformities are the result of nonsynostotic causes (i.e., positional plagiocephaly) and which are associated with a true absence of a cranial vault suture (i.e., craniosynostosis) is critical to establishing a diagnosis and formulating the appropriate treatment plan. The importance of a correct diagnosis is underscored by the dramatically different treatment approach to each condition. Craniosynostosis requires surgical treatment, whereas benign positional skull molding (positional plagiocephaly) is not associated with any neurologic consequences and typically requires conservative nonsurgical management using custom-made cranial orthotic devices or in the case of torticollis, neck physical therapy.
A careful history and clinical examination are the primary basis upon which the diagnosis of craniosynostosis is made. Because craniosynostosis is an intrauterine event, parents will often report that the deformity was noted immediately after birth and never improved after that time. By contrast, the typical history provided in cases of positional plagiocephaly is that the child’s head was well rounded at the time of birth and the asymmetry was noted 3 to 6 months later, suggesting postnatal deformation of a flexible cranial vault. Clinical examination of the infant with craniosynostosis is a key element in establishing the diagnosis. Each form of craniosynostosis will produce a unique dysmorphology that resulted from arrested skeletal growth perpendicular to the affected suture and continued or compensatory growth at the sutures that remain open. This combination of arrested growth in one region and additional growth in others may explain how unilateral sutural involvement results in a bilateral deformity and how fusion of one of the sutures within the posterior cranial vault will produce a deformity that affects the entire anteroposterior (AP) dimension of the cranium. An examiner familiar with these aberrant growth patterns and the characteristic dysmophologies specific to each type of synostosis will be able to consistently establish an accurate clinical diagnosis.
The diagnosis of craniosynostosis is made primarily on the basis of a careful clinical examination, but must be confirmed radiographically. Following clinical evaluation by a pediatric craniofacial surgeon and/or pediatric neurosurgeon, a complete skull series of plain radiographs is obtained to evaluate the cranial vault sutures. Standard views of the cranial vault are often all that is required to establish the absence of a cranial vault suture and confirm the diagnosis of craniosynostosis. In cases where all of the sutures cannot be adequately visualized using plain films, additional imaging with the use of CT is indicated. In addition to confirming the diagnosis, high quality craniofacial CT scans provide more detailed 3-D morphologic information, which is useful during the surgical planning phase. One-millimeter axial and coronal cuts with overlap and 3-D reconstructions of the cranial vault, cranial base, orbits, and maxillofacial skeleton are recommended. Precise patient positioning is important in obtaining a diagnostic scan, and this may require sedation or general anesthesia when the patient is an infant or young child.
Although technologic advancements have dramatically increased the accuracy of standard radiographic studies and CT imaging, incorrect and outdated terminology is still occasionally used in describing cranial vault sutures. At some medical centers, radiologists and surgeons continue to use terms such as “fibrous synostosis,” “suture dysfunction,” and “impending synostosis” when describing cranial vault sutures that otherwise appear open. We now know that craniosynostosis is in the vast majority of cases an intrauterine event that is either present at birth or not at all. Therefore if a suture appears patent on a radiographic study that is of diagnostic quality, the diagnosis of craniosynostosis is ruled out. Although the postnatal fusion of cranial vault sutures has been described in the scientific literature, it is considered an extremely rare event that is specifically related to specific clinical and syndromic conditions. The use of the terms described earlier is not consistent with contemporary knowledge about craniosynostosis, may be confusing to parents and health care providers, and may even result in inappropriate surgical treatment for patients who do not have craniosynostosis.
TREATMENT OF NONSYNDROMIC CRANIOSYNOSTOSIS
GENERAL CONSIDERATIONS
There are two primary objectives in the contemporary surgical management of nonsyndromic craniosynostosis: first, the release of the involved (i.e., fused) suture so that brain growth can proceed in an unrestricted fashion and second, the reconstruction of all dysmorphic skeletal components so that a more anatomically correct shape is achieved. The best results are achieved with collaboration between a pediatric craniofacial surgeon and pediatric neurosurgeon.
Early surgical techniques used to treat craniosynostosis involved only the removal of involved sutures via a “strip craniectomy. ” Generally, these limited craniectomy procedures would be performed by a neurosurgeon working independently. The theory behind this approach was that release of the suture would allow unrestricted brain growth and that the expanding brain would adequately recontour the bones without the need for formal craniofacial reconstruction. Although this approach does allow for cerebral decompression, the reality is that a dysmorphic skull will not reshape itself, even in the presence of an expanding brain, and the result is a residual bony deformity.
Modern surgical management of craniosynostosis involves release of the involved suture(s) with a formal craniotomy performed by a neurosurgeon. At the same time, reconstruction requires the removal, dismantling, and reassembly of all dysmorphic skeletal components into a more anatomically correct position. Depending on which specific cranial vault suture is affected, the reconstruction may also involve the orbits. The exact surgical plan is formulated based on the extent of the skeletal deformity, the suture or sutures involved, and the age of the patient at the time of diagnosis. In most cases of nonsyndromic, single-suture craniosynostosis, one operation to simultaneously release the suture and reshape the affected skeletal parts will adequately address the problems definitively. This is more likely when the surgical procedure is carried out during the first year of life and the patient has normal brain growth. In addition to a combined craniofacial and neurosurgical approach, involvement of a number of other pediatric subspecialists, including pediatric anesthesiologists and pediatric intensivists, is required.
In recent years, the use of endoscopic instrumentation in the surgical management of craniosynostosis has been described and shows promise in the management of specific patients. An endoscopically assisted approach is used to carry out a strip craniectomy for the release of the involved suture(s) and the creation of multiple “barrel-stave” osteotomies within the cranial vault. The primary advantage of this technique is a much smaller surgical incision. Other described theoretic advantages of an endoscopic approach have included the elimination of blood transfusions, less morbidity, and decreased length of hospitalization. The main disadvantage encountered with the use of an endoscopic procedure is that the surgical maneuvers performed do not address the cranial and/or orbital dysmorphology. Instead, the child’s cranial vault dysmorphology is addressed with the use of molding helmets during the postoperative period. Full-time molding using a custom helmet (i.e., cranial orthotic) is often required for 8 to 12 months following the surgical procedure. Residual cranial vault deformity is a frequent problem. Because the successful use of custom-made cranial orthotic devices or helmets depends upon ongoing brain growth, children diagnosed with craniosynostosis after 6 months of age are not considered good candidates for endoscopic treatment. Another consideration is the idea that endoscopic surgery is “nonsurgical” or “minimally invasive.” Although the length of the incision used is significantly decreased when the endoscopically assisted procedure is used, significant complications, including hemorrhage, laceration of the sagittal sinus, the need for blood transfusion, dural tears, and brain injury, are possible. Blood transfusion is required in a significant proportion of children undergoing the operation. Surgeons contemplating an endoscopic procedure must be aware of these potential problems and make standard preparations (e.g., type and cross blood, secure neurosurgical support, etc.) to deal with them as necessary.
TIMING OF SURGERY
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


