Fig. 5.1
Pictorial descriptions of different types of polymeric nanoparticles and their respective functionalities. Surface modifiers, targeting molecules (ligands) and incorporated agents are shown
5.2.3 Liposomes
Nano-sized liposomes can be prepared using the rehydration technique, followed by extrusion. Generally, known amounts of lipids, with and without cholesterol, are dissolved in a low-boiling organic solvent such as ethanol at 60 °C. Using a rotary evaporator, the organic solvent is evaporated off to form a thin film. This is then hydrated with an aqueous solution containing the drug or protein. Rehydration leads usually to the formation of multilamellar vesicles (MLV), which have sizes in the range of 0.5–5 μm. These are downsized to about 70–500 nm (unilamellar vesicle, or ULV) by extrusion through membrane filters at high pressures [9] to produce liposomes with incorporated drugs. In the above example, the drug is incorporated by the ‘passive’ loading method. For more hydrophilic drugs, an ‘active’ loading method is required to incorporate therapeutically meaningful quantities. For passive loading, a lipophilic drug is dissolved in the organic phase, while a hydrophilic drug is dissolved in the aqueous phase. An example of an active method is the use of a pH gradient [10] or an electrochemical gradient [11, 12] for different drug types.
5.2.4 Dendrimers
Dendrimers are essentially hyperbranched polymer structures that can potentially ‘encapsulate’ drugs or proteins. The control of release is exerted primarily through diffusion and in some cases by degradation. These hyperbranched structures (polymers) are prepared by a very specific reaction sequence, usually starting with an amine-terminated molecule. Such a molecule is reacted with an acrylate ester and then subsequently with ethylene diamine to yield a ‘full-generation’ dendrimer [13]. Repetition of the above reactions yields a highly branched structure with internal ‘cavities’ that may hold metal atoms or other guest molecules by virtue of the presence of amine groups. Molecules may be conjugated to the ‘interior’ groups as well as the ‘surface’ groups, and these molecules may be a drug, peptide, antibody or PEG. Conjugation generally opens up possibilities for selective targeting of tissue but seldom for sustained release applications.
5.3 Clinical Applications
5.3.1 Cancer Chemotherapy
We now review the applications of the above classes of nanoparticulate carriers in different therapies. By far, the greatest attention has been paid to targeting tumour tissue as nanoparticles have the ability to traverse easily out of blood vessels into tumour tissue in comparison to their microparticulate cousins. In addition, surface modification of nanoparticles for evasion of the reticuloendothelial system (RES) as well as for penetration of selected tissue is no less feasible for nanoparticles. So in what follows, we discuss the relative successes of the different particle types in targeting tumour tissue.
Active and Passive Targeting
Two concepts have been used for targeting cancer tissue. Passive targeting involves injectable drug carriers that have been surface modified (Fig. 5.1) to evade the RES such that their blood lifetime is relatively long. Long-lived particles have a much greater chance of reaching the blood vessels surrounding solid tumours, and then extravasate by virtue of their size. Once in tumour tissue, the relative lack of lymphatic drainage allows for slow release of the payload into the surrounding tissue. The whole effect goes by the name of enhanced permeability and retention (EPR).
Active targeting relies on conjugating a targeting ligand (usually an antibody) to the surface of the particle such that the ligand targets cancerous tissue only. Most of the work to date has focussed on folate and transferrin receptor targeting; these two receptors are over-expressed in cancerous cells. Others have tried to exploit the presence of a tissue-specific antigen (TSA) whose antibody may be used for the targeting. In this approach, the nanosize is not as critical, although it still helps in facile extravasation.
Liposomes
Approved Products
Liposomal delivery systems have been by far the most successful of the nanoparticulate carriers. No fewer than four pharmaceutical products that use liposomes as a carrier have been approved: of these, Doxil® was approved in 1995 for the treatment of ovarian and other cancers. Other approved liposomal products include DaunoXomeTM, LipoDox and Myocet, while another liposomal formulation (Synergene Therapeutics) for activating the p53 (tumour suppressor) gene has successfully completed Phase 1 trials. In the discussion below, we will focus on the development of Doxil and comment on the shortcomings and advantages of liposomal nanocarrier systems.
Doxil
The story of successful development of the very first nanoliposmal carrier is both long and intriguing [14]. The story includes many discoveries wrapped into one product: for example, the idea of ‘pegylation’ of the carrier to increase blood circulation lifetimes and the concept of ‘active drug loading’ into the core of the liposome were both crucial to successful development. The important features of this so-called stealth liposome are depicted in Fig. 5.2. The lipid molecule, which is distearoyl-phosphatidyl-ethanolamine (DSPE), is conjugated at its amine end to a 1,900-MW mPEG molecule [15]. Doxorubicin (Dox) is partially crystallized in the aqueous core (Fig. 5.2), and the control of its release through the membrane is accomplished mostly by the crystallization and somewhat by incorporation of cholesterol in the membrane: the cholesterol tends to make the membrane more rigid and thus slow down diffusion. Most of the drug (~12 % by weight of lipid) is claimed to be inside the aqueous core of the liposomes. Such high loadings are generally only possible with what are called active loading methods. This uses a salt such as ammonium sulfate inside the core of the liposome to bind to the drug molecule as drug sulphate and precipitate which then drives the influx of drug to the liposome core [12].
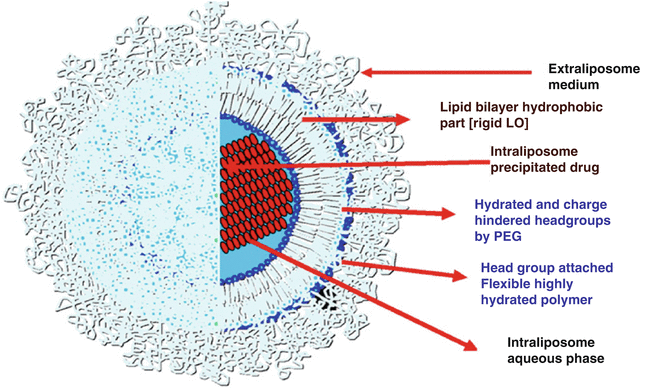
Fig. 5.2
Pictorial description of “stealth” liposome: Doxil (Reprinted from Barenholz [14]. With permission from Elsevier). LO ‘liquid ordered’ and PEG ‘polyethylene glycol’ represents different domains of Doxil
The liposomal drug thus formulated works on the principle of ‘passive targeting’. Upon intravenous (IV) administration, the carrier (with incorporated drug, Dox) circulates for about 30–45 h without being phagocytosed. During this period, very little of the Dox is released into the bloodstream: hence, controlling (minimizing) the release for at least 45 h is crucial to the success of this mode of drug administration. The long blood circulation time of the pegylated carrier is partly responsible for accumulation at the tumour site: statistically, the chance of particles accumulating near the tumour tissue is high because of the number of blood vessels feeding it. The size of the liposomal particles (80–100 nm) [16] is the second reason for accumulation into tumour tissue by virtue of ‘extravasation’ or leakage through the highly permeable capillaries surrounding tumour tissue. It is now generally accepted that extravasation is possible only for particles less than 600 nm in size. Clinically, this is highly significant because accumulation in liver and spleen is considerably reduced for the stealth particles, and this translates directly to tumour size reduction and its maintenance over 100 days compared to free drug injections and thus to a substantial improvement in the survival rate of mice with colon carcinoma.
Doxil® was a pioneer in the field of passive targeting. Its success prompted several imitations, each of which touted advantages over Doxil®. One example is Myocet®, which claimed an even higher loading of Dox (~25 %) in particles of about 190 nm. Interestingly, this product consists of non-pegylated egg PC/cholesterol lipids and appears to minimize one side effect of Doxil®, which is the so-called hand-foot syndrome [17], that is exacerbated with long-circulating carriers. Myocet® is approved in Europe for treating metastatic breast cancer (co-administration with Herceptin) and is being considered for approval in the USA. Unlike Doxil®, which is stored as a suspension, Myocet® is a lyophilized powder that is reconstituted prior to IV administration.
Liposomal Systems in Pre-clinical and Clinical Phases
Other cancer targets (besides simply killing tumour cells) have been addressed. One such is activation of the tumour suppressor gene p53, especially in lung cancer. Early attempts to do so were reported by researchers at the M. D. Anderson Cancer Center in Houston, with successful suppression of primary and metastatic lung tumour growth [18] in animal studies. Co-authors included employees of Introgen, which subsequently seemed to have abandoned liposomal delivery in favour of adenoviral carriers for delivering the gene [19]. Since then, however, Introgen appears to have folded due to lack of funding.
As mentioned above, another company that has reported clinical trials with a liposomal vector is SynerGene Therapeutics based in Washington D.C., USA. From various company reports, this appears to be a liposomal vehicle with targeting ligands, presumably a folate receptor–targeting ligand. The payload is likely to be a p53 gene also. A patent [20] describes an ‘immunoliposome’ which is based on the liposome component DOPE (dioleyl phosphatidyl ethanolamine) incorporating a transferrin receptor–targeting ligand. The immunoliposome is complexed with p53 wild-type genes and targets the transferrin receptor, which is over-expressed in many tumours.
Polymeric Nanomicelles
Liposomes are relatively narrow distribution in their sizes following extrusion. The sizes are generally not very stable against both aggregation and fusion or disassembly over time; these are self-assembled structures that are in a state of thermodynamic equilibrium at the temperature of interest and with the requisite concentration of lipids. The self-assembling nature confers shape, stability and uniformity, however, unlike solid nanoparticles. Similarly, spherical polymeric nanomicelles in fact can spontaneously form under the appropriate conditions in aqueous or non-aqueous media depending on the molecular structure and extent of amphiphilic character.
In terms of papers and patents, nanomicellar drug delivery has been a fertile field. For polymeric molecules to self-assemble to spherical micelles or lamellar structures, block co-polymers are ideal, with one block being hydrophilic and the other hydrophobic. Triblocks, diblocks and even four-armed blocks have been used [21, 22]. Relative to liposomes, success in the clinic has been harder to come by, but there are some notable exceptions, as we will see in the next section.
The key to the success of a polymeric nanomicelle carrier is CMC [23]. Although micelles are prepared at concentrations that ensure they are the predominant species, upon injection (IV) into the body, there is immediate and substantial dilution, which may lead to disassembly of the micelles. Hence, an effective micellar carrier has a very low CMC to ensure that the self-assembled structure is retained following administration. In general, the CMC is influenced by these factors:
1.
The hydrophobic–hydrophilic ‘mismatch’: the greater the difference between the two groups forming the micellar molecule, the lower the CMC
2.
The lengths of the two segments: in general, the longer these segments, the lower the CMC
Usually above the CMC, spherical micelles are generated, although other structures are possible and have been observed. For drug delivery, spherical carriers that maintain their shape over time of storage are preferred for stability of release of drugs. Drug partitioning is also enhanced when the drug hydrophobicity is matched with that of the hydrophobic segment. In general, the lower the CMC, the lower the amount of free molecules in the ‘suspension’, as illustrated in Fig. 5.3.
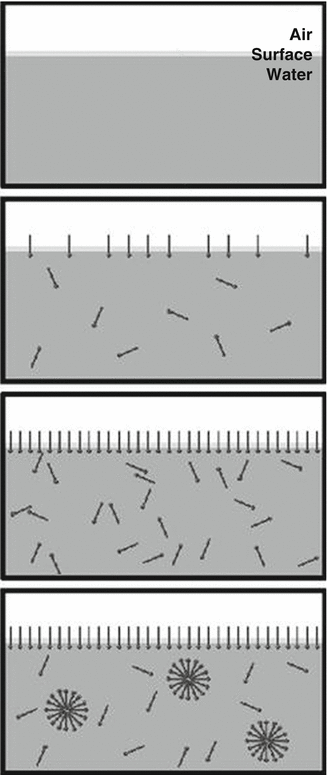
Fig. 5.3
Pictorial depiction of the formation of micelles by addition of amphiphilic molecules to water (Reprinted from Critical micelle concentration [23].with permission from Creative Commons Attribution)
Clinical Candidates
The use of nanomicelles for passive targeting to certain tumours has been spearheaded by Japanese researchers at the National Cancer Center /Research Institute East in Chiba, Japan, in conjunction with the University of Tokyo. The approach is to encapsulate the generally hydrophobic drug candidates in the micellar core and manipulate release predominantly through degradation of the hydrophilic segment. Equilibration of drugs between the core and the hydrophilic segment is discouraged via low partitioning.
Years of exploratory work have led to the development of three promising nanomicellar formulations, paclitaxel (NK105), doxorobucin (NK911) and cisplatin (NC-6004). The statuses of these products are briefly covered below.
NK105
The NK in the name of this experimental product is due to ‘Nippon Koyaku”, the Japanese company that is developing and financing the clinical trials. The polymer is a diblock copolymer of PEG and modified polyaspartate [24] where half of the aspartate groups are converted to the more hydrophobic derivative, 4-phenyl 1-butanolate. As explained above, this derivatization enhances the hydrophobicity of the segment and leads to a lower CMC. The overall MW of the polymer used was 20,000, of which the PEG block was 12,000 and the aspartate block was 8,000. Approximately 20 % of the polymer weight can be loaded with paclitaxel held in the micellar core by hydrophobic self-association. The micelles obtained in water or aqueous media were lyophilized to obtain particles which upon reconstitution yielded micelles of average diameter about 85 nm, with size ranging from 20 to 430 nm.
Paclitaxel is a potent anti-cancer drug that suffers from poor bioavailability due to its low solubility. In addition, systemic injection with a solubilizer such as Cremaphore EL induces hypersensitivity reactions. Moreover, peripheral neuropathic reactions as well as neutropenia have been reported with repeated use of paclitaxel [25]. For all these reasons, a better delivery system that can at least partially target tumours is highly desirable for paclitaxel. Following promising animal data, a Phase 1 trial was conducted in 2007 on 19 patients [26] who had solid tumours refractory to conventional chemotherapy. NK105 showed slow clearance, with a half-life of about 5–6 h (compared to about 1 h for injected paclitaxel), thus enabling passive targeting to tumours. Neuropathy, which is a common side effect of paclitaxel, was not observed. Hypersensitivity was also negligible, even without co-administration of steroids. Since then, a Phase II (efficacy) study has been completed, and a Phase III study in breast cancer patients [27] is under way. While the timeline has been a long one, it appears that NK105 is likely to be approved for use in certain cancers.
NC-6004
Based on early work at the University of Tokyo [28], the company NanoCarriers (NC) has developed a micellar delivery system for another cancer drug, Cisplatin. Cisplatin (a Pt (II) complex with cis-dichlorodiamine) is an anti-cancer drug that suffers from various limitations, including nephrotoxicity and neuropathy, with repeated use [29]. The micellar system using PEG and poly(glutamate) has been developed to incorporate cisplatin via complexation of the glutamate unit to Pt(II) [30, 31]. This is a novel way to improve cisplatin loading and control its release. In fact, the in vitro release of cisplatin into saline is triggered by chloride substitution for glutamate in the complex and shows no burst but sustained release over 150 h as shown in Fig. 5.4. The size of the particles remains stable, at about 30 nm over 72 h, and presumably decreases beyond that due to micellar dissociation into unimers.
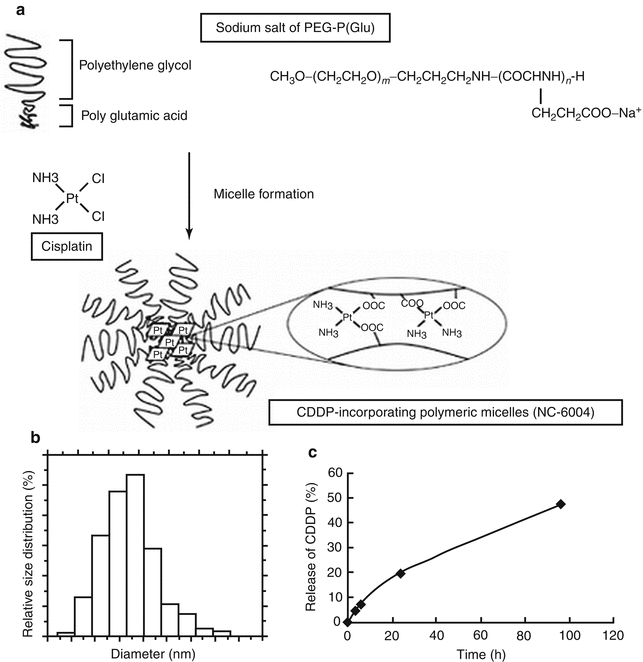
Fig. 5.4
Schematic of cisplatin structure and its incorporation into a micelle via complexation with a polyamino acid (polyglutamate). The hydrophilic polymer, PEG, also acts as anti-phagocytic layer, similar to pegylated liposomes (Reprinted from Uchino et al. [93]. With permission from Nature Publishing Group). (a) Chemical structures of CDDP and PEG–P(Glu) block copolymers, and the micellar structures of CDDP-incorporating polymeric micelles (NC-6004). (b) The particle size distribution of NC-6004 measured by the dynamic light-scattering method. The mean particle size of NC-6004 was approximately 30 nm. (c) Release of CDDP from NC-6004 in saline at 37°C.
Clearance and tissue distribution are superior for the micellar formulation of cisplatin compared to free cisplatin. In the mouse study [30], tumour accumulation over 24 h is roughly 5–10 fold higher than free cisplatin depending on tumour location. Tumour inhibition was demonstrably superior with the micellar formulation for mice bearing colon adenocarcinoma. As of now, this compound is in Phase III trials in Asia and in Phase II in the USA. It is expected to win approval in a couple of years.
NK911
A micellar formulation for Dox has been developed as well, and it is interesting to compare micellar Dox against liposomal Dox. The micellar system for Dox is similar to that for paclitaxel (NK105), utilizing the same PEG–aspartate copolymer (PEG MW = 5,000; polyaspartate has 30 repeats) [32]. The Dox in this case is conjugated to the aspartate moiety for greater loading efficiency. The conjugated drug confers sufficient hydrophobicity to enable formation of stable micelles. This enhanced hydrophobicity allows more packing of ‘incorporated’ Dox (as distinct from ‘conjugated’ Dox). The conjugated Dox is not active and is not released from the micelle, while the incorporated Dox is released slowly over 24 h. The mean diameter of the micellar particles is 42 nm after freeze-drying of the micellar solution.
An interesting comparison with Doxil (the liposomal Dox) shows the micellar Dox to be cleared 400 times faster than Doxil [33]. However, it is not clear that this lower blood lifetime necessarily translates into lowered accumulation in tumour tissue as the volume of distribution for Doxil appears higher than that for micellar Dox. Another study [34] showed that in vitro, the Doxil formulation was more stable to leakage of drugs than the NK911, which also points to potentially greater side effects caused by free Dox in systemic administration. It is likely that for these reasons, development of NK911 has been discontinued.
Other Nanomicelle Systems and Studies
There is an excellent review of the large amount of research in the field of fully degradable micellar nanocarriers [35]. The major polymeric systems studied appear to be block copolymers of hydrolyzable polyesters with PEG. The hydrolyzable polyesters include poly (caprolactone) (PCL) [36, 37], PLA [21, 22] and PLGA [38]. In addition, ‘stealth’ entities other than PEG have been studied as micelles, including Poly(vinyl pyrrolidone) or PVP and PVA. However, no other entity has been shown to work as well as PEG.
Previous studies have reported the optimum PEG length and configuration needed for stealthiness [39]. In our study, we synthesized triblock PLA-PEG-PLA copolymers (ABA type) as well as PEG-PLA-PEG copolymers (BAB type) with varying lengths of PEG and PLA and measured uptake by blood cells in vitro. We also compared the uptake behaviour of triblocks with diblocks of PEG-PLA. The lower the uptake, the greater is the ‘stealthiness’ or longer the blood lifetime. Confocal microscopy shows that as the size of the nanomicelle increases, the phagocytic uptake increases compared to the smaller particle. In general, uptake was greater for ABA triblocks compared to diblocks and BAB triblocks of similar PEG and PLA lengths; in addition, it was found that the PEG segment length had to be above 5,000 MW in order for any effect to be seen for uptake reduction by blood cells. Surface concentration of PEG was not the only determinant factor for reducing uptake; the surface conformation of the PEG was important as well. More free PEG molecules (i.e. with conformational freedom) exerted a bigger effect compared to more constrained molecules.
Loading of hydrophobic drugs into the micelle is generally easier than the loading of hydrophilic drugs. As noted above, paclitaxel has been loaded to about 20 % in a typical micelle. However, control of release in the clinically tested formulations is still inadequate. In our own work, we find that this control of release can be improved substantially [21, 22] by the use of the relatively hydrophobic polyesters, which have the added advantage of full biodegradability into harmless products.
Loading of more hydrophilic drugs and control of its release requires special techniques, including drug conjugation. Fortunately, most of the anti-cancer drugs tend to be fairly hydrophobic. This is in fact the main reason that these aqueous micelles have been predominantly studied for anti-cancer therapy. Loading of antibiotics, for example, will not be high, and loaded drugs may release quicker than other drug types. One example of hydrophilic drug loading is in the example shown above for cisplatin using poly glutamate as a conjugating agent (NC6004). Cisplatin is moderately hydrophilic, and the loading can be increased to 35–40 % by conjugation. Although other approaches have been proposed including the so-called core surface-cross-linked micelles [40], it is not clear that the increased loading is without an accompanying increase in release rate.
Solid Nanoparticles
These are nanoparticles that are composed mainly of polymers that are not fluid at 37 °C but have glass transition temperatures (T g) or melting points (T m) above 37 °C. Of the polymers evaluated to date, PLGA is the most versatile in that the properties can be fine tuned by varying the ratio of lactide to glycolide in the copolymer: for example, the T g may be varied between 45 and 70 °C in changing the ratio from 50 % of l-lactide to 85 % of l-lactide. The copolymers are fully biodegradable, and the degradation products are relatively harmless. The overall degradation rate may also be manipulated by changing the lactide-to-glycolide ratio. In this section, we will focus on PLGA nanoparticles as applied to cancer therapy.
Solid nanoparticles are defined as matrix formulations whereby the drug is either dissolved or dispersed in a polymer that is processed into a nanoparticulate form (other than via self-assembly), whereas nanocapsules are core–shell particles made with two different types of polymers or with lipids as cores and polymeric shells. Of the two, it appears that nanocapsules are difficult to fabricate and most core–shell particles tend to have micron dimensions. Thus, most of the discussion below relates to matrix-type nanoparticles. The main advantages of using these particles in preference to micelles and liposomes are simplicity of manufacture, greater stability and better control over release of drugs. Disadvantages are relative difficulty in attaching PEG or ligands to the particle. As mentioned above, matrix microparticulate drug delivery systems have been around for a long time, but nanoparticulate ones are rare. As far as we are aware, none of the Food and Drug Administration (FDA)–approved nanosystems is based on solid nanoparticles.
PLGA nanoparticles can be made using a variety of ways, from emulsion techniques to spray-drying. The emulsion technique is preferred with the use of ultrasound for size reduction. In a variation of the emulsion technique called the spontaneous emulsification solvent diffusion (SESD) method, a mixed solvent system is used for the polymer solution, which is then emulsified into an aqueous medium containing a surfactant. To minimize agglomeration of the particles, a low-MW PVA surfactant is used, and two completely water-miscible solvents can be used for the polymer phase [41]. This technique produces non-aggregating particles in the size range 100–200 nm that remains stable even after freeze-drying.
Such particles can incorporate a wide range of drug molecules, from low-MW hydrophobic molecules [42] to proteins [43] and even plasmid deoxyribonucleic acid (DNA) [44]. As mentioned earlier, such nanoparticles may again be surface modified: PLGA has also been coated with PEG and PEG-like molecules including the surfactant molecule PEG-PLA-PEG [45]. Grafting of PEG molecules to surfaces of already formed nanoparticles is seldom reported, most of the ‘chemically bound’ PEG nanoparticles relying instead on copolymerization with PLA [46] or PLGA [47] or even polycyanoacrylate [48].
Passive Targeting
In spite of several years of active research following the approval of the first PLGA microsphere formulation (Zoladex) in 1989, nanoparticulate PLGA, decorated with surface-bound PEG, has not been shown to be superior to either liposomes or micelles for passive targeting to tumours. The reasons for this are twofold: (a) the PLGA nanoparticles tend to aggregate over time, and lyophilization and reconstitution does not always produce particles of the same size range, and (b) although drug loading is increased with these solid NPs, release is retarded due to diffusional barriers. Too much retardation is not useful in passive targeting as slow release at the tumour leads to sub-cytotoxic levels of drugs.
Active Targeting
Given the lack of advantage in passive targeting, most of the PLGA nanoparticle work has concentrated on the so-called active targeting approach. In this approach, a ligand is attached to the particle that binds to receptors or molecules that are unique to cancerous cells while carrying their cargo of cytotoxic drugs. The most studied ligand is the folate ligand that binds to folate receptors that are usually over-expressed in cancerous cells, particularly in epithelial malignancies such as breast cancer and endometrial cancer. The concept is to attach the folate ligand covalently to the PEG molecule usually with a spacer such as PEG. An example of this approach is found in Liang et al. [49]. In this example, a new terpolymer had to be synthesized which contains blocks of PLGA, PEG and folate. Incomplete conjugation of folate at one end leads to some level of ‘stealthiness’ (Fig. 5.5) due to the ‘free’ PEGs on the surface.
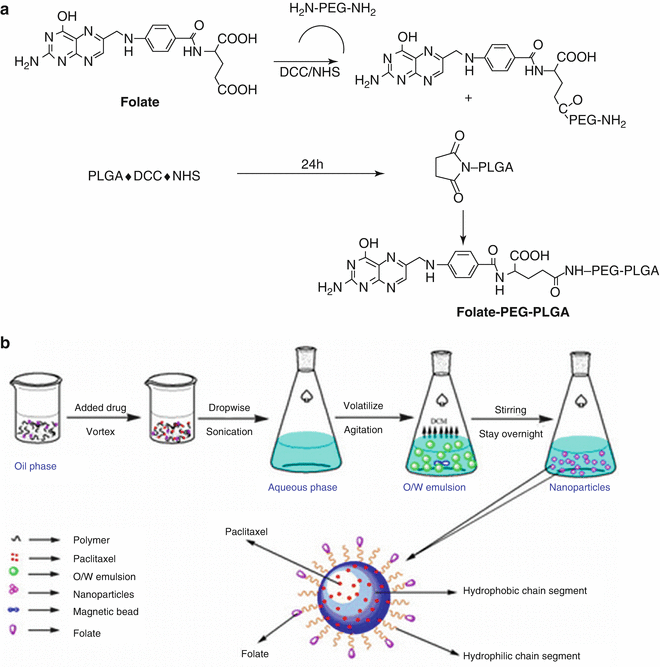
Fig. 5.5
Schematic of synthesis of folate-decorated PLGA-PEG nanoparticles (Reprinted from Liang et al. [49]. With permission from Elsevier). (a) Structure of FOL–PEG–PLGA. (b) Schematic representing formulation of FOL–PEG–PLGA nanoparticles and the process for drug loading
The nanoparticles generated by volatilization of the organic solvent from the emulsion were about 220 nm in average diameter and a surface charge of −8 mV with the folate added. Using in vitro studies, it was found qualitatively that the folate-conjugated particles were internalized to much greater extent by human carcinoma cell lines when compared to PLGA-PEG particles. Incorporating paclitaxel into these particles and then injecting (via the tail vein) into mice bearing endometrial cancer cells showed greater tumour volume reduction for the folate-conjugated nanoparticles. Many more such examples can be found in the literature. In spite of encouraging animal data, none of these ligand-bearing nanoparticles have been tested in humans. It does appear that ‘stealthiness’ and targeting are mutually incompatible features of a nanoparticle. If sufficient stealthiness is achieved by PEGylation, generally the targeting capability is compromised, and vice versa. The best solution therefore appears to be local administration of the NPs rather than intravenous administration.
Comparing Nanoliposomes, Nanomicelles and Solid Nanoparticles for Drug Delivery
Stability Issues
In general, nanoliposomal carriers appear to be more stable than nanomicelles, particularly to drug leakage. For cancer applications, it is critical that substantial leakage of drugs must not occur during the period of carrier circulation in blood (i.e. within the half-life in blood). Doxil, for example, sustains the release of drugs in ambient conditions to much beyond 45–50 h so that the drug remains encapsulated until its encounter with tumour cells.
One explanation for this greater stability to leakage is given by Lasic [50]. Lasic argues that liposomes do not form unilamellar or multilamellar structures spontaneously, unlike micelle formation from block copolymers. This means that the lamellar structures are not thermodynamically favoured. Yet, paradoxically, once they are formed (by an extrusion process, i.e. external energy input), these structures tend to be more stable due to what Lasic calls ‘kinetic trapping’. This kinetic trapping is essentially caused by hydration of the polar heads, and thus it results in lack of penetration of the lipid cores by water. This stability is crucial to clinical success especially if intravenous injection is used to administer the formulation.
As noted above, micelles are thermodynamically favoured structures for block copolymers in solvent (i.e. good solvent for one of the constituents). The stability of these micelles, however, is dependent on the actual magnitude of the CMC: the lower the CMC, the greater is the tendency for these micelles to form and, of course, greater the resistance to structure breakdown upon dilution. Achieving sufficiently low CMCs is a challenge. Since micellar formulations are naturally diluted upon injection into the bloodstream, it is speculated that they tend to become monomeric entities upon injection; this leads to immediate drug release. Another complication is that added salts generally increase CMCs and hence break down existing micelle structures. Liposomes are not susceptible to this as the kinetic entrapment ensures that dilution has very little effect on the liposome. In other words, water penetration into the core of a liposome is not enhanced by adding more water, hence the apparent stability of liposomes following injection. Incorporation of cholesterol in liposomal formulations is also believed to enhance this kinetic stability.
Solid nanoparticles do not have leakage issues when stored at ambient temperature. However, nanoparticles aggregation over time is a significant issue. Additionally, heterogeneity of shape (and size) appears to be more of a problem with nanoparticles, with consequent variability in drug release rates. These have precluded their widespread use in cancer therapy to date.
Blood Lifetimes
In general, whichever entity has greater stability in blood will probably have enhanced blood lifetimes, greater tumour tissue accumulation (although this one can be influenced by other factors such as size and distribution in specific tissues), better control of drug release and enhanced bioactivity. Stability in this context also refers to stability against phagocytosis by elements of the RES. There is widespread agreement that the nano-sized particles have greater blood lifetimes than micron-sized ones; furthermore, that attachment of PEG molecules confers enhanced resistance to phagocytosis for these nanocarriers. There is experimental evidence that nanoliposomes exhibit longer blood lifetimes than nanomicelles of comparable size. Thus, it appears that the ‘presentation’ of the PEG corona to the phagocytic cells is not the same for liposomal PEG and micellar PEG. Exactly why this is so has not been clarified yet.
For solid nanoparticles, the PEG must be covalently attached to the matrix polymer via an extra synthesis step. In principle, this should work just as well for solid NPs as for liposomes and micelles. However, the heterogeneity in shape/size perhaps contributes to a more rapid clearance of PLGA particles as compared to liposomes. For non-cancer applications, however, blood lifetimes are not critical, but control of drug release is, as we will see in the examples that follow.
5.3.2 Ocular Applications
Sustained delivery of therapeutic agents is desirable for many ocular diseases, including glaucoma and macular degeneration, both chronic conditions without any known cures. However, the impermeability of the corneal epithelium and rapid clearance by tears preclude the use of topical eye drops for sustained delivery in spite of several years of research. Drugs applied via eye drops penetrate the barriers in the front of the eye at the best by about 5 % of dose; the dosing lasts for about 20–30 min on average. For the diseases to be discussed below, longer-duration drug action is highly desirable both from an efficacy as well as a patient compliance standpoint.
Glaucoma
Glaucoma is a chronic, progressive optic neuropathy that causes irreversible blindness. It is the major cause of irreversible blindness worldwide. The global burden of glaucoma is estimated to rise to affect 80 million worldwide by 2020 primarily due to an increasing ageing population in the world. Elevated intraocular pressure (IOP) is the only known effective modifiable risk factor. The use of daily eye drops containing hypotensive agents to lower the IOP remains the first-line treatment for glaucoma. Like any chronic therapy, ocular hypotensive agents require patient adherence. In addition, patients have trouble using eye drops and applying them correctly in the eye. A consequence to poor patient adherence is treatment failure and a poor outcome from disease progression. It is estimated that at least 10 % of blindness is directly attributed to poor patient adherence to prescribed medications.
Clearly, eye drops cannot sustain IOP lowering for more than 24 h at best. To achieve sustained duration of IOP lowering, other modes of administration are needed. One approach is to use a ‘punctual plug’ which effectively plugs the tear duct; if an IOP-lowering drug is incorporated into the body of the plug, it may be released slowly over time. The company Ocular Therapeutics has reported the development and testing in humans of such a plug containing the drug travoprost [51]. Although such a mode of administration [52] (Fig. 5.6) offers promise for longer-duration therapy, it is more invasive than a sub-conjunctival injection that can also sustain the duration of IOP lowering for at least 3 months following a single injection. For this, nanoliposomes have been found to be successful [53]. The challenge is to incorporate sufficient amounts of the drug into the nanoliposomes such that a single 150 mL injection contains sufficient drug to last over 3 months; secondly, the release of drug must be slow enough to sustain IOP lowering over the same time period. The nanosize helps to reduce opacity in the anterior chamber as well as not to be irritating to the patient.
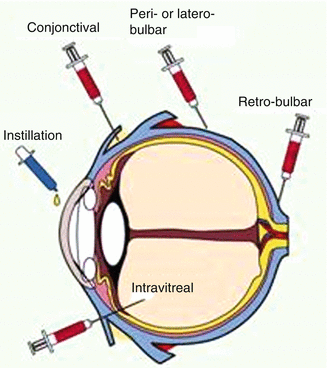
Fig. 5.6
Routes of administration of nanocarriers in the eye (Courtesy of Claudia Di Tommaso)
Work in our group showed that the prostaglandin analogue, latanoprost, may be loaded up to 10 % by weight of lipids in liposomes [54] and the release sustained over 2 months in vitro. In vivo studies in diseased monkeys showed duration of IOP lowering for up to 4 months. There were no deleterious local effects of the sub-conjunctival injection: the bleb that is formed initially resolves in 24 h. This approach opens up possibilities for sustained therapy of anterior chamber diseases that are chronic in nature. The required dosing over 3 months is usually high for most drugs, and so a suitable nanocarrier system must be identified that can incorporate such large doses without losing shape or aggregating.
Posterior Segment Diseases
One site of action where sustained delivery capability is needed is the posterior eye segment. There are several conditions that require sustained therapy: diabetic oedema, macular degeneration (the so-called wet kind) and infections, including cytomegaloviral infections. Topical administration is not a feasible option due to the impermeability of the various barriers. Currently, all the three conditions are treated with either intra-vitreal implants or intra-vitreal injections. Both procedures have associated risk factors, such as retinal detachment and vitreous haemorrhage. Furthermore, such procedures also suffer from poor patient acceptance. Thus, sustained delivery options for the back of the eye are an important area of research and have been for some years.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


