where,
-
D h is the hydrodynamic diameter
-
D t is the translational diffusion coefficient
-
k B is Boltzmann’s constant
-
T is thermodynamic temperature
-
η is dynamic viscosity
In a DLS experiment, a laser beam is directed at the nanoparticle dispersion, and fluctuations in the intensity of the scattered light are monitored with a photon detector and related to the size of a hypothetical hard sphere that diffuses in the same fashion as the nanoparticles being measured. Thus, the equivalent-sphere hydrodynamic diameter obtained from DLS provides no information about the shape of the nanoparticles. The size distribution of the nanoparticle dispersion, as indicated by the polydispersity index (PDI), can also be obtained from the DLS measurement. The larger the PDI, the broader is the size distribution, and a PDI value from 0.1 to 0.25 indicates a narrow size distribution [10]. DLS is a very popular technique for measuring the size of nanoparticles because it requires minimal sample preparation and can be rapidly carried out (a few minutes per run) with small volumes of dilute dispersions. However, the results obtained from DLS can be skewed by the presence of aggregates or dust since the intensity of the light scattered by the nanoparticle varies as the sixth power of its diameter. Thus, for DLS measurement, it is critical to ensure that the nanoparticles are well dispersed. The usefulness of DLS is also limited if a multimodal particle size distribution is present. For example, when a mixture of 20 and 100 nm nanoparticles is measured, the signal of smaller particles is lost because the scattering intensity of small particles is masked by that of larger particles [10]. The concentration of the dispersion may also affect the hydrodynamic size, as illustrated by Lim et al. using magnetic nanoparticles without surface coating dispersed in deionized water [11]. A sample that is too dilute may not result in sufficient scattering events for a proper measurement, while multiple scattering can occur in a highly concentrated sample. Furthermore, nanoparticles at high concentration have a higher tendency to aggregate. In view of the sensitivity of DLS to the presence of aggregates, this technique is useful for monitoring colloidal stability. This technique has been used by a number of investigators to assess the stability of nanoparticles in physiologically relevant media [12–14].
Electron microscopy can be carried out to complement the DLS technique for particle size measurements. Scanning electron microscopy (SEM) utilizes a high-energy electron beam to scan over the surface of the sample. The X-rays, backscattered electrons, and secondary electrons resulting from the interaction of the sample with the electron beam are collected and converted to provide information about the sample, including its surface features, size and shape of the features, and composition. The scanning electron microscope operates at high vacuum, and the specimen must be electrically conductive or sputter coated with a conductive layer (e.g., platinum). For SEM characterization of nanoparticles, the dry sample can be mounted directly on a sample holder. In conventional SEM, the electron beam is emitted from an electron gun with tungsten or LaB6 filaments. By replacing these with a field emission gun, field emission scanning electron microscopy (FESEM) with higher resolution can be achieved. Transmission electron microscopy (TEM) also utilizes an electron beam to interact with the sample under high vacuum, but in this case, the transmitted electrons are detected. TEM provides higher resolution than SEM and provides more information at the atomic scale such as the crystal structure, but the sample must be thin enough to allow electron penetration [4]. For TEM characterization of nanoparticles, a dispersion of the nanoparticles is usually deposited directly onto support grids or films.
Sample preparation for electron microscopy may introduce artifacts, e.g., the dehydration of colloidal nanoparticles can affect the structure and morphology of the sample [15]. Some nanoparticles such as liposomes or polymers are invisible to TEM without heavy metal staining (e.g., with phosphotungstic acid) since they do not deflect the electron beam sufficiently [4]. As such, particle sizes determined by TEM and DLS can differ substantially. This is illustrated by a comparison of these two methods for measuring the size of magnetic nanoparticles either coated with oleic acid or grafted with a hydrophilic polymeric coating [16]. The TEM image of the oleic acid–stabilized magnetic nanoparticles dispersed in hexane shows that these nanoparticles are nearly monodispersed with an average diameter of about 12 nm (Fig. 3.1a). This value is close to the mean hydrodynamic diameter of ~15 nm as determined by DLS. The presence of electron-transparent oleic acid chains (chain length ~2 nm [17]) on the surface of the magnetic nanoparticles may have contributed to the slightly larger size as determined by DLS. In the case of magnetic nanoparticles grafted with hyperbranched polyglycerol (HPG) and methotrexate (MTX), the TEM image (Fig. 3.1b) indicates similarity in size with the oleic acid–stabilized magnetic nanoparticles even though its magnetic core constitutes less than 35 % of the total weight since in both cases TEM only shows the electron-dense magnetic nanoparticle core. On the other hand, the hydrodynamic diameter of these nanoparticles has increased to ~32 nm, and this significant increase in hydrodynamic diameter is attributed to the swelling of the hydrophilic HPG shell in water during the DLS measurement.
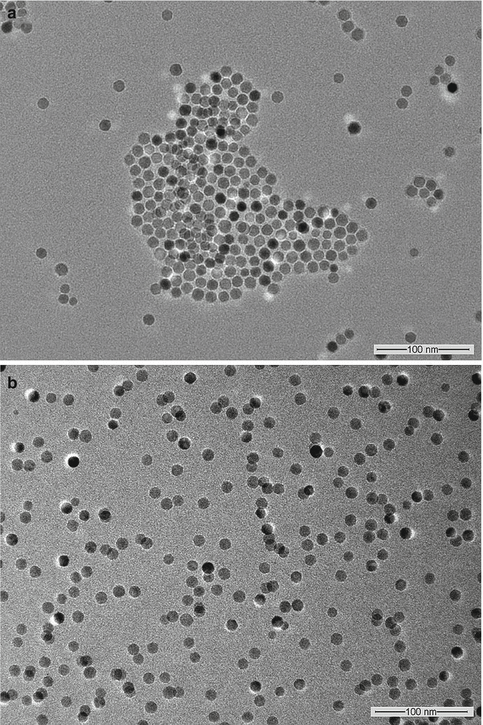
Fig. 3.1
TEM images of (a) oleic acid-stabilized magnetic nanoparticles dispersed in hexane, and (b) magnetic nanoparticles grafted with HPG and MTX dispersed in water (Reprinted from Li et al. [16]. With permission from Elsevier)
3.2.2 Surface Charge
Surface charge is another factor that plays an important role in determining how nanoparticles interact with proteins and cell membranes and their subsequent uptake into the cells [18, 19]. Furthermore, the surface charge of nanoparticles also affects their colloidal stability since it directly impacts the electrostatic repulsion of nanoparticles in dispersion [20]. The surface charge of a nanoparticle in dispersion affects the distribution of ions in the surrounding interfacial region, resulting in an increased concentration of counterions near the surface. The liquid layer surrounding the particle exists as two parts: the Stern layer, which is the region closest to the surface, where the ions are considered immobile, and an outer region that allows diffusion of ions. In this surrounding electric double layer, there is a notional boundary (slipping plane) within which the liquid moves together with the particle. The potential measured at this slipping plane is the zeta potential, which is not exactly the surface charge but is the potential of practical interest in dispersion stability because it determines the interparticle forces [21].
Zeta potential can be determined using electrophoretic light scattering (ELS), also known as laser Doppler electrophoresis or laser Doppler velocimetry. In this experiment, an electric field is applied across a colloidal dispersion. Charged particles in the dispersion will move toward the electrode of opposite polarity (electrophoresis) with a velocity or electrophoretic mobility that is proportional to the zeta potential. A laser beam is incident on the dispersion, and the electrophoretic mobility can be determined from the frequency shift of the light scattered by the moving particles. The measured electrophoretic mobility (U E) is converted into zeta potential (ζ) through Henry’s equation [22]:
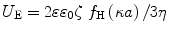
where,
-
ε is the dielectric constant of the dispersant
-
ε 0 is the vacuum permittivity
-
f H(κa) is the Henry’s function
-
η is the viscosity
The dimensionless product κa describes the ratio of the particle radius (a) to the “thickness” of the double layer (κ −1). At large κa (~100), the equation can be simplified to the Smoluchowski approximation with f H(κa) = 1.5 [23]. On the other hand, when κa << 1, f H(κa) = 1, and this is known as the Hückel approximation. Doane et al. have provided a contour plot based on Ohshima’s approximation to Henry’s formula identifying the Smoluchowski and Hückel regimes for different nanoparticle sizes and solution ionic strengths, as shown in Fig. 3.2 [22].
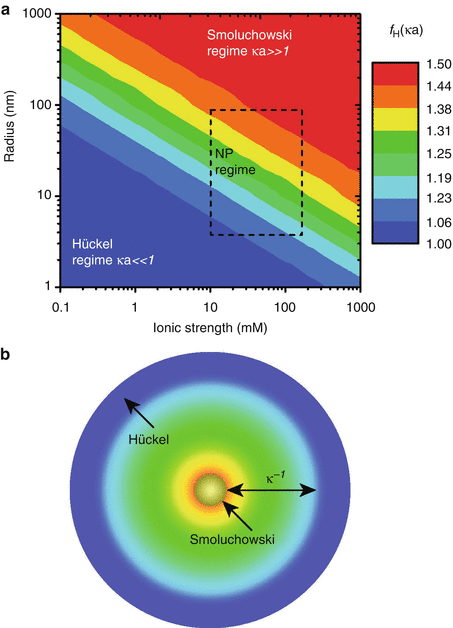
Fig. 3.2
(a) Henry’s factor f H(κa) colored according to the analyte radius and solution ionic strength (both with logarithmic scales): the Smoluchowski [f H(κa) = 3/2] and Hückel [f H(κa) = 1] limits are identified by red and blue, respectively, with the rectangle identifying particle radii and ionic strengths prevailing in common nanoparticle applications. (b) The Debye layer thickness κ −1 colored according to the accompanying Henry factor f H(κa) in (a) (Reprinted from Doane et al. [22]. With permission from American Chemical Society)
A number of commercial instruments are available for the measurement of zeta potential. While the measurement is easily carried out with a dispersion of the nanoparticles, the effect of temperature, ionic strength, and pH of the dispersion on zeta potential has to be recognized [10, 24, 25]. A 5 °C variation in temperature can lead to significant changes in zeta potential [25]. High ionic strength decreases the electric double layer, resulting in a reduction of the absolute value of zeta potential, and divalent ions have a stronger effect than monovalent ions. At low pH, the nanoparticles acquire more positive charges, and the zeta potential decreases as the pH is increased. Thus, it is crucial to provide precise information on the dispersion when reporting zeta potential measurements.
3.2.3 Surface Composition
Nanoparticles possess a very high surface area–to-volume ratio, and the importance of the nanoparticle surface property and chemistry in determining their suitability for specific bioapplications cannot be underestimated. For example, there has been increasing interest in the use of superparamagnetic iron oxide nanoparticles for biomedical applications since these nanoparticles have low toxicity and are suitable for in vivo applications as they are biodegradable, with the iron product being recycled by cells. But monodispersed superparamagnetic iron oxide nanoparticles are often synthesized via a high-temperature decomposition process where oleic acid is used to stabilize the formed product [26, 27]. As a result, the nanoparticles disperse well in organic solvents but not in aqueous media. For biomedical applications, the surface of these nanoparticles has to be modified with a hydrophilic and biocompatible coating, and depending on the desired application, ligands may also be bound on the nanoparticle surface to promote interactions with cellular receptors. Thus, analysis of the surface composition of the nanoparticles is needed to confirm the success of the tailoring process.
X-ray photoelectron spectroscopy (XPS), also known as electron spectroscopy for chemical analysis (ESCA), is one of the most widely used techniques for analyzing the surface composition of nanoparticles. The popularity of XPS stems from its ability to (a) identify and quantify the elemental composition of the outer 10 nm or less of any solid surface for all elements from lithium to uranium on the assumption that the element of interest exists at > 0.05 atomic %. Hydrogen and helium are not detectable due to their extremely low photoelectron cross sections, (b) reveal the chemical environment where the respective element exists, and (c) obtain the above information with relative ease and minimal sample preparation [28]. In XPS measurement, the sample is placed in a chamber under ultrahigh vacuum and subjected to x-rays, most commonly Al Kα x-rays (photon energy = 1486.7 eV). An electron is ejected from an atomic energy level by the X-ray photon, and its energy is analyzed by the spectrometer. By measuring the kinetic energy of the emitted photoelectrons, E K, and knowing the photon energy hν, the binding energies of the photoelectrons, E B, can be calculated using the Einstein equation:
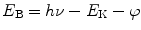
where φ, the work function of the instrument, can be determined by calibration. It can be seen from the above equation that only binding energies lower than the exciting radiation can be probed.
Since the binding energies of the electron orbitals in atoms are known, the positions of the peaks in the XPS spectrum can be used to identify the atomic surface composition of the sample. Each element has a characteristic electronic structure and thus a characteristic XPS spectrum. Tables of binding energies for identifying and interpreting XPS spectra are available in the literature [29]. The usefulness of XPS for characterizing nanoparticles can be illustrated by using this technique for monitoring changes in the surface composition of magnetic nanoparticles subjected to surface modification, as shown in Fig. 3.3 [16]. Oleic acid–coated magnetic nanoparticles were first grafted with 3-(trimethoxysilyl)propylmethacrylate (MPS), and HPG was then grafted on the MPS-modified magnetic nanoparticles via thiol-ene click reaction. Figure 3.4a shows the wide scan or survey scan obtained at low resolution of oleic acid–coated magnetic nanoparticles. The C 1 s peak associated with oleic acid, Fe 2p and Fe 3p peaks arising from the magnetic nanoparticles, and O 1 s peak from both oleic acid and magnetic nanoparticles can be identified. Figure 3.4b shows the corresponding spectrum of these nanoparticles after surface grafting with HPG. The increase in the O 1 s signal relative to the C 1 s signal is in line with the higher O/C ratio of HPG compared to oleic acid. The S 2p signal at a binding energy of about 168 eV (Fig. 3.4b) arises from the grafting of thiolated HPG via thiol-ene click reaction on the magnetic nanoparticles [16]. Thus, a comparison of Fig. 3.4b with Fig. 3.4a provides supporting evidence that HPG has been grafted on the surface of the magnetic nanoparticles. The wide scan identifies the elements present, and quantification of the chemical composition as percentage atomic concentrations is possible from the peak area of each element corrected by the relative sensitivity factors (specific to the instrument) [30].
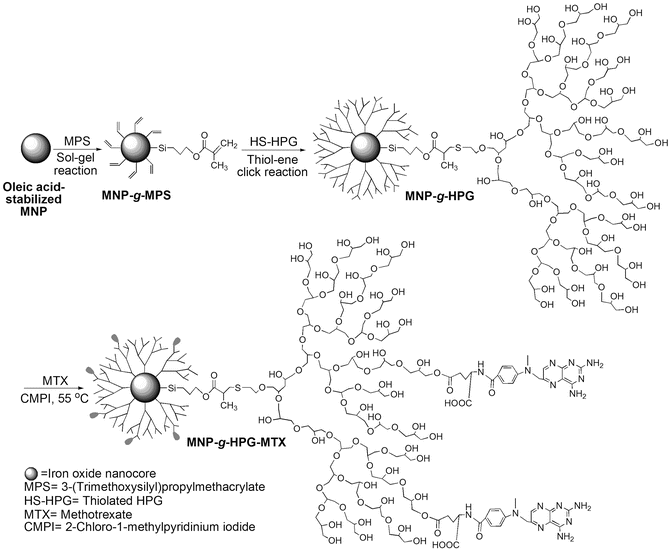
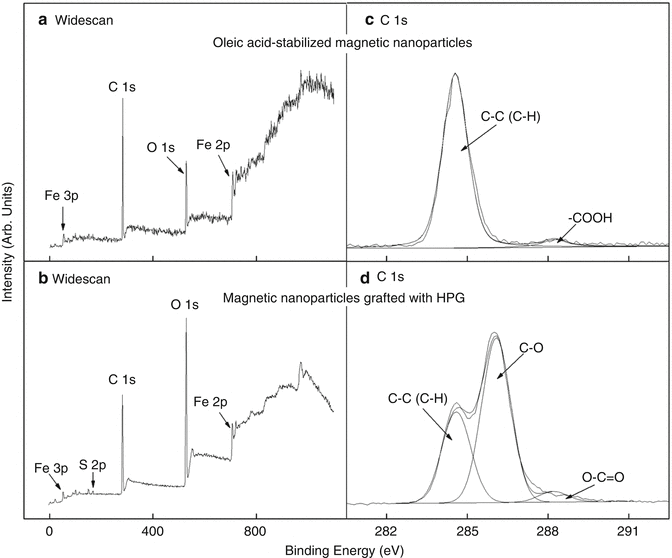
Fig. 3.3
Schematic illustration of the synthesis of HPG-grafted magnetic nanoparticles with conjugated MTX (Reprinted from Li et al. [16]. With permission from Elsevier)
Fig. 3.4
XPS widescan spectra of (a) oleic acid-stabilized magnetic nanoparticles, and (b) magnetic nanoparticles grafted with HPG, and C 1 s core-level spectra of (c) oleic acid-stabilized magnetic nanoparticles, and (d) magnetic nanoparticles grafted with HPG
XPS not only allows the identification of the elements constituting the sample but also provides information on their chemical state based on the binding energy shift (or chemical shift). For example, the C 1 s high-resolution core-level spectrum of the oleic acid–coated magnetic nanoparticles in Fig. 3.4c shows a dominant peak at 284.6 eV attributable to C-C/C-H groups and a much smaller peak at 288.4 eV attributable to the COOH group of oleic acid. The shift of the COOH peak to a higher binding energy relative to the C-C/C-H peak arises from a decrease in electron density in the C valence shell due to bonding to oxygen, which results in an increase in the binding energy of the C 1 s core orbital. The ratio of the peak areas gives the relative proportion of each component on the surface. In Fig. 3.4c, the area ratio of the C-C/C-H peak to the COOH peak is calculated to be 17.6:1, which is close to the theoretical ratio of 17:1 expected from the chemical structure of oleic acid. In Fig. 3.4d, the C 1 s peak envelope arising from C being present in multiple environments is more complex than in Fig. 3.4c, where two peak components can be clearly delineated. The peak envelope in Fig. 3.4d can be deconvoluted into three-component peaks at 284.6, 286.1, and 288.3 eV with the same full width at half maximum (FWHM). The peak at 284.6 eV is assigned to C-C/C-H groups as in Fig. 3.4c, while the dominant peak at 286.1 eV is attributed to C-O groups, consistent with the chemical structure of HPG. The minor O-C=O peak is likely to be from the MPS anchoring groups which did not react during the thiol-ene click reaction.
XPS is clearly a powerful technique capable of identifying a wide range of elements and their chemical states on surfaces. The analysis can be readily carried out, but the sample must be compatible with the ultrahigh vacuum environment. Since XPS is a surface-sensitive technique, cleanliness during sample preparation and during analysis is imperative to obtain the true surface composition. It must also be emphasized that although peak deconvolution and peak fitting tools are commercially available, curve fitting of peak envelopes must be undertaken with caution especially in the presence of overlapping peaks. Poorly fitted spectra with arbitrariness in peak width and shape can lead to incorrectly interpreted XPS data. More detailed discussions on curve fitting of core-level XPS data and the importance of combining curve fitting information with other supporting information and experiments are available in the literature [28, 31].
Auger electron spectroscopy (AES) is another common surface analysis tool for nanoparticles. Both AES and XPS detect electrons emitted from samples with kinetic energies typically below 2,000 eV, but in AES, the sample is irradiated with electrons instead of x-rays [32]. AES and XPS provide similar information, but AES gives a higher lateral resolution since the electron beam can be focused to a smaller size than x-rays. However, the electron beam can also impart more damage to the sample surface than x-rays. Baer et al. have provided a comparison of the types of information that can be extracted from nanoparticles using the surface analysis methods, AES, XPS, time-of-flight secondary-ion mass spectrometry (TOF-SIMS), low-energy ion scattering (LEIS), scanning tunneling microscopy (STM), and atomic force microscopy (AFM), as shown in Table 3.1. The ion-based techniques, TOF-SIMS and LEIS, can provide information on surface chemical composition to complement the electron spectroscopic techniques, but the scanning probe microscopy techniques, STM and AFM, are more suited for extracting information on physical properties like size, morphology, and surface heterogeneity.
Table 3.1
Types of information that can be extracted from nanoparticles using surface analysis tools, with several characteristics of the methods
|
Information available
|
Probe
|
Detected
|
Lateral resolution
|
Information depth
|
Depth resolution
|
|
|---|---|---|---|---|---|---|
|
Electron spectroscopies
|
||||||
|
Auger electron spectroscopy (AES)
|
Surface composition of individual large nanoparticles or distribution of smaller nanoparticles (depending on spatial resolution of specific instrument)
Enrichment or depletion of elements at surface
Presence and/or thickness of coatings and/or contaminants
|
Electrons (~3 to 20 keV)
|
Auger electrons
|
10 nm
|
~10 nm
|
~2 nm
|
|
X-ray photoelectron spectroscopy (XPS)
|
Analysis of a collection of particles deposited on a substrate or other support
Surface composition and chemical state
Presence and nature of functional groups on the surface
Enrichment or depletion of elements at surface
Presence and/or thickness of coatings or contaminants
Nanoparticle size (when smaller than ~10 nm, can sometimes determine average particles size when too small to be detected by other methods or in complex matrix)
Electrical properties of nanoparticles and coatings
|
X-rays
|
Photoelectrons
|
~2 μm
|
~10 nm
|
~2 nm
|
|
Incident ion methods
|
||||||
|
Time-of-flight secondary-ion mass spectrometry (TOF-SIMS)
|
Usually analysis of a collection of particles or larger individual particles deposited on a supporting substrate
Presence of surface coatings or contaminants on collections of nanoparticles
Functional groups on surface
|
Ions (~3 to 20 keV)
|
Sputtered ions
|
~50 nm (inorganic);
>200 nm (organic)
|
~1 nm (inorganic);
~1 nm (organic)
|
~1 nm (inorganic);
~10 nm (organic)
|
|
Low-energy ion scattering (LEIS)
|
Presence of ultra thin coating or contamination
Effects of size
|
Ions (~2 to 10 keV)
|
Elastically scattered ions
|
~100 μm
|
~10 nm
|
~0.2 nm
|
|
Scanning probe microscopies
|
||||||
|
Scanning tunneling microscopy (STM)
|
Electrical characteristics of individual nanoparticles
Nanoparticle formation and/or size distribution of particles deposited or grown on a surface
|
Stylus
|
Tunneling current
|
~1 nm
|
~10 nm
|
|
|
Atomic-force microscopy (AFM)
|
Shape, texture and roughness of individual particles and their distribution for an assembly of particles
When particles structure is known, can provide information about crystallographic orientation
|
Stylus
|
Force or displacement
|
~1 nm
|
~10 nm
|
|
Fourier transform infrared spectroscopy (FTIR) has been used for identifying functional groups on nanoparticles. In this technique, infrared radiation is passed through a sample, and some of the radiation is absorbed by the sample. The resulting spectrum shows a series of absorption bands characteristic of the frequency of vibrations of bonds between the atoms making up the sample. Since each sample comprises a unique combination of atoms, the spectrum provides a distinct identification of the sample akin to that of a molecular “fingerprint.” The region between 2.5 and 25 μm (4,000–400 cm−1) is most frequently used in modern infrared instruments [33]. The preparation of solid nanoparticle samples for FTIR analysis usually involves the milling of the sample with KBr and then compressing the fine powder into a thin pellet which is placed in the sample holder of the FTIR spectrometer. KBr is transparent to IR radiation and hence will not interfere with the absorption spectrum. However, since KBr is hygroscopic, precaution must be taken to ensure that the KBr is dry otherwise absorption bands from the adsorbed water may interfere with the sample’s spectrum.
The use of FTIR for identifying the functional groups on nanoparticles is illustrated using the example of HPG-grafted magnetic nanoparticles discussed above. The FTIR spectrum of this sample shows dominant absorption bands at about 3,000–3,500 cm−1 attributed to the stretching vibrations of –OH groups, at 2,926 and 2,871 cm−1 due to CH2 stretching vibrations, and at 1,086 cm−1 associated with the stretching vibrations of C–O–C ester groups (Fig. 3.5a). These groups are characteristic of the HPG graft layer. The HPG-grafted magnetic nanoparticles can be modified to be a drug delivery system for targeting cancer cells by conjugating MTX on the nanoparticle surface. MTX is one of the most widely used chemotherapy drugs for the treatment of many types of cancers [34, 35]. Furthermore, since MTX is an analogue of folic acid and the folate receptor is overexpressed on many types of human cancer cells, the MTX-functionalized nanoparticles can potentially act as a cancer-targeting agent in addition to being an anticancer drug [36]. The FTIR spectrum of the MTX-functionalized nanoparticles (Fig. 3.5b) can be compared with those of the HPG-grafted nanoparticles before conjugation with MTX (Fig. 3.5a) and free MTX (Fig. 3.5c). The doublet absorption bands at 1,646 and 1,605 cm−1, which are the characteristic peaks of the amide groups of free MTX, appear in the spectrum of the MTX-functionalized nanoparticles but not the HPG-grafted nanoparticles, thus confirming the successful conjugation of MTX in the former.
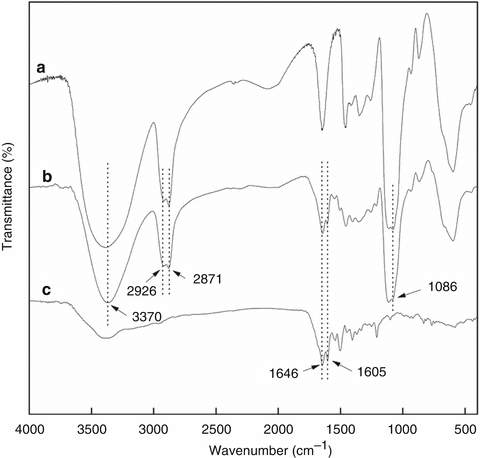
Fig. 3.5
FT-IR spectra of (a) magnetic nanoparticles grafted with HPG, (b) magnetic nanoparticles grafted with HPG and MTX and (c) free MTX
In addition to providing identification of functional groups on nanoparticles, FTIR can also be used for quantitative analysis. For quantitative analysis, calibration curves will first have to be established. Shan et al. used FTIR to quantify the amount of hemoglobin encapsulated in polymeric nanoparticles fabricated by the double emulsion method from poly(ethylene glycol)–poly(lactic acid)–poly(ethylene glycol) triblock copolymers [37]. Polyacrylonitrile (PAN) was chosen as an internal standard, and a constant amount was introduced to the calibration and test samples in order to avoid errors arising from the environment and laboratory manipulation. The calibration samples comprise hemoglobin, PAN, and blank polymeric nanoparticles. The IR absorbances at 1,540 cm−1 of hemoglobin (amide II) and at 2,241 cm−1 of PAN (−C≡N) were measured to establish the calibration equations relating the absorption band height ratio of hemoglobin and PAN as a function of the weight ratio of hemoglobin and PAN. From the absorbances of hemoglobin and PAN measured in the spectra of the test samples, these equations were then used to calculate the amount of encapsulated hemoglobin.
Although FTIR in the transmission mode is the most common, attenuated total reflectance–Fourier transform infrared spectroscopy (ATR-FTIR) has also been widely used. Unlike transmission FTIR, in which the IR beam travels in a straight path through a sample, in ATR, the infrared beam is directed into a high–refractive index crystal where it undergoes total internal reflection. The reflected IR beam creates an evanescent wave, which projects into the sample placed in contact with the crystal. Some of the energy of the evanescent wave is absorbed by the sample, and the attenuated radiation is passed to the detector. Tsai et al. used modified ATR-FTIR to carry out a quantitative study of competitive molecular adsorption on nanoparticles [38]. In these experiments, Au nanoparticles were deposited from a dispersion to form a film onto the germanium ATR crystal of the FTIR spectrometer. A flow cell was attached to the top of the ATR crystal, solutions with various concentrations of molecular adsorbates were then introduced into the flow cell, and the IR spectrum was collected. The surface density of the adsorbed molecules on the Au nanoparticles was computed by comparing the signal intensity of the IR band related to the adsorbate with the calibration curve established using the unbound molecule in deionized water.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


