Figure 15.1. Schwannoma (hematoxylin-eosin, 203). This tumor is encapsulated and shows typical palisaded nuclei surrounding acellular areas (Verocay bodies).
Schwannomas are fairly common in the head and neck and can elicit pain and other symptoms. They are typically well circumscribed and often encapsulated. The histologic hallmark of schwannomas is the presence of a cellular “Antoni A” pattern (Figure 15.1) with palisading nuclei (Verocay bodies) and an “Antoni B” pattern with a hypocellular loose stroma. Marked cellular atypia may be present in degenerated or “ancient” schwannomas, although mitoses are virtually absent.
Neurofibromas present as solitary (circumscribed), diffuse, or plexiform lesions. They are composed of spindle cells with wavy nuclei arranged either haphazardly, in short fascicles, or in a storiform pattern. The stroma contains varying amounts of wire-like wavy collagen and myxoid areas. Solitary neurofibromas are cured by conservative excision.
Syndromic association with multiple BPNST can occur as follows: neurofibromas in neurofibromatosis (NF) type 1, schwannomas in NF type 2, mucosal neuromas in multiple endocrine neoplasia syndrome type 2B (Figure 15.2), and perineuriomas in NF type 2, although these are exceedingly rare.
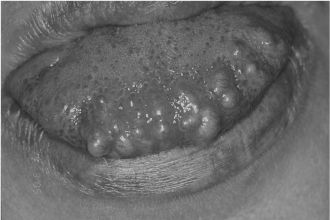
Figure 15.2. Multiple neuromas in multiple endocrine neoplasia 2B syndrome presenting as multiple submucosal nodules.
15.1.4 Bisphosphonate-associated osteonecrosis of the jaws
Bisphosphonate-associated osteonecrosis is defined as bone necrosis in patients with a history of previous or current bisphosphonate therapy who have not received radiotherapy in the involved area. It is much more common in i.v forms of therapy, due to which osteoclastic activity is severely compromised. Clinically, bisphosphonate-associated osteonecrosis may arise de novo or secondary to odontogenic infections or invasive dental procedures. Patients present with exposed necrotic bone and may develop pain, abscess formation, and pathologic fracture (Figure 15.3). There is no proven cure thus far. Treatment can include cessation of bisphosphonate therapy followed by debridement (depending on the patient’s medical condition and overall prognosis), vascularized grafts, prevention of acute infections, and palliative care. Patients under other forms of bone-targeted therapy, such as receptor activator of nuclear factor-κB ligand inhibitors, may also develop osteonecrosis of the jaws, clinically indistinguishable from bisphosphonate-associated osteonecrosis.
15.1.5 Cemento-osseous dysplasia
Also known as osseous dysplasia, cemento-osseous dysplasia (COD) is a common benign fibro-osseous disease of the alveolar processes of the jaws. COD is classified into three distinct clinicoradiologic entities: periapical, focal, and florid. Periapical COD by definition involves the periapex of anterior mandibular teeth. It has a marked predilection for women, and is more common in black patients. Focal COD is confined to a single site of involvement, typically in the posterior mandible. Florid COD involves multiple foci and may cause slight expansion of the cortices. In early stages of disease, all three forms of COD present as asymptomatic radiolucencies on conventional radiographs and computed tomography, gradually becoming more calcified. Eventually, lesions may become completely sclerotic and hypovascular, and hence prone to necrosis after trauma or odontogenic infections. CODs may be associated with the formation of simple bone cysts (SBCs). Histologically, CODs are composed of cementum-like calcifications and irregular islands of trabecular and woven bone set in a moderately cellular fibrovascular stroma (Figure 15.4). In cases where lamellar bone is prominent, CODs can be mistaken for chronic osteomyelitis. Unlike other calcifying bone lesions, CODs normally do not require any treatment, unless in the presence of secondary osteomyelitis.
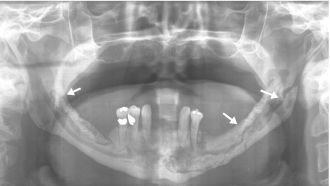
Figure 15.3. Bisphosphonate-associated osteonecrosis of the mandible. Panoramic radiograph showing a complex and open pathologic fracture involving the entire mandible (arrows).
Familial gigantiform cementoma is a very rare autosomal dominant disorder characterized by multifocal fibro-osseous lesions of the jaws, leading to dramatic expansion of the cortices and disfigurement. It is regarded by some as a part of the COD disease spectrum because of similar histopathologic features and occurrence in the tooth-bearing area of the jaws. Partial excision leads to recurrence or secondary osteomyelitis. Therefore, extensive resection and reconstruction may be required.
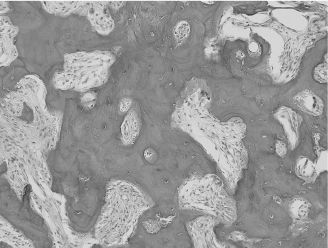
Figure 15.4. COD (hematoxylin-eosin, 203). Interconnecting trabeculae of woven bone in a moderately cellular and fibrovascular stroma. This case was diagnosed clinically as periapical cemental dysplasia.
15.1.6 Central giant cell granuloma
Central giant cell reparative granulomas (CGCGs) are benign osteolytic lesions composed of osteoclastic giant cells, fibrous tissue, extravasated red blood cells, and reactive bone formation (Figure 15.5). They have a predilection for the mandible and may be associated with significant expansion and osteolysis. Histologic features are not predictive of clinical behavior. Furthermore, because CGCGs are histologically identical to brown tumors of hyperparathyroidism, the latter must be excluded clinically. The histologic differential diagnosis also includes cherubism, ABC, and giant cell-rich variants of osteosarcoma. Again, clinicopathologic correlation is required. Treatment of small lesions consists of conservative curettage, whereas large lesions may respond to intralesional injections of corticosteroids or alpha-interferon in combination with intranasal calcitonin therapy.
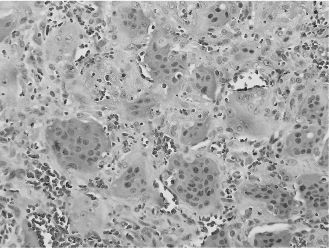
Figure 15.5. CGCG (hematoxylin-eosin, 403). Densely packed multinucleated giant cells obscuring the fibrovascular background stroma.
Giant cell tumors are common tumors of the long bones, but their occurrence in the jawbones is a controversial topic. They are histologically comparable to CGCG, to a point where they may be impossible to differentiate from aggressive types of CGCG. In general, giant cell tumors are benign but behave more aggressively than CGCGs.
15.1.7 Central ossifying fibroma
Central ossifying fibroma (COF), also known as cemento-ossifying fibroma, is a rare neoplasia of the jawbones with a predilection for the posterior mandible of female patients. Eventually, if left untreated, COFs may cause significant expansion of the cortices and dental displacement. In early stages of disease, radiographic features may be indistinguishable from those of focal COD or osteoblastoma. Histologic features can resemble those of other benign fibro-osseous lesions, although COFs tend to show osteoblastic rimming and varying levels of mineralization. An important distinguishing feature is that the lesions are easily separated from surrounding bone, as opposed to focal COD or fibrous dysplasia, both of which merge with adjacent bone. Complete enucleation is the treatment of choice.
Juvenile active ossifying fibroma is an aggressive variant of COF that is seen in young patients, typically in the maxilla or paranasal sinuses. Histologic features include a cellular stroma containing spindle cells and giant cells, strands of immature osteoid with numerous osteoblasts, and variable amounts of psammomatoid calcifications. Recurrences are much more common than in conventional COF, thereby justifying complete resection.
Hyperparathyroidism-jaw tumor syndrome is a very rare autosomal dominant disease characterized by parathyroid tumors, renal lesions, uterine polyps, and occasional multicentric fibro-osseous lesions of the jaws that resemble COFs.
15.1.8 Cherubism
A rare autosomal dominant inherited disease, cherubism is characterized by osteolytic fibrous lesions resulting in bilateral enlargement of the jaws (Figure 15.6). It occurs in the first decade of life and tends to stabilize at puberty, at which point lesions may evolve into mature bone. Histologic features include cellular fibrous stroma in which are scattered varying amounts of multinucleated giant cells, extravasated red blood cells, and hemosiderin. Perivascular hyalinization is a distinguishing feature (Figure 15.7). Treatment is the topic of ongoing debate and varies according to the extent of cosmetic or functional involvement.
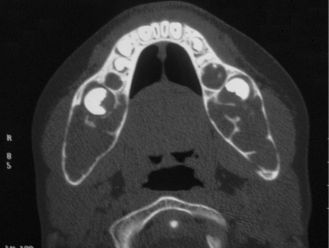
Figure 15.6. Cherubism. Axial computed tomogram showing bilateral and symmetric expansion of the mandibular cortices.
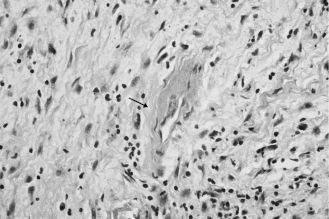
Figure 15.7. Cherubism. Perivascular hyalinization (arrow) and fibrous stroma (hematoxylin-eosin, 403). Other areas (not shown here) contained numerous multi-nucleated giant cells.
15.1.9 Chondrosarcoma
Chondrosarcoma is one of the most common malignancies of bone. In the head and neck area, the most frequent site of occurrence is the larynx, followed by the jaws, the maxillary sinus walls, the nasal cavity, and the base of the skull. Most cases arise centrally within bone, but they may also occur in periosteal locations from preexisting benign cartilaginous tumors. Incidentally, any intraosseous lesion of the jaws that produces cartilage should be viewed with extreme suspicion; such lesions are malignant until proven otherwise. Well-differentiated chondrosarcomas (grade 1) are composed of rather uniform cells whereas poorly differentiated tumors (grade 3) are characterized by pleomorphic tumor cells, necrosis, and a lesser amount of chondroid matrix. Clear cell and dedifferentiated variants have been described. Prognosis and treatment depend on histologic grade. Treatment ideally consists of complete resection with uninvolved margins. The efficacy of conventional radiotherapy is not clearly established, although proton therapy is showing promise.
A mesenchymal chondrosarcoma is a related malignancy composed of a dense population of small, round cells with a hemangiopericytoma-like vasculature. Cartilaginous differentiation can be bland and minimal. Mesenchymal chondrosarcomas of the jaws seem to have a better prognosis than nongnathic tumors. However, overall prognosis is worse than for conventional chondrosarcomas.
15.1.10 Chordoma
Chordomas are relatively rare malignant neoplasms that recapitulate embryonic remnants of the notochord. In the head and neck area, they have a tendency to occur in the base of the skull and may present with various symptoms depending on their exact location. The periphery of these tumors is often well demarcated by a pseudocapsule. Histologic examination of chordomas typically reveals “bubbly” or vacuolated epithelioid cells (physaliphorous cells) arranged in chords and pseudoglandular structures. The stroma is markedly mucoid, which confers a gelatinous consistency to the tumor. The treatment of choice may include complete resection and combined proton radiotherapy. The 5-year survival rate is distinctly poorer after age 40.
15.1.11 Craniopharyngioma
Craniopharyngiomas are benign, slow-growing but aggressive epithelial tumors thought to be derived from remnants of Rathke’s pouch. Although most of these tumors occur in the suprasellar or intrasellar region, they may also occur in ectopic locations such as the nasopharynx and sphenoid bone. There are two histologic types: adamantinomatous (by far the most common) and papillary squamous. Adamantinomatous craniopharyngiomas are composed of ameloblastoma-like epithelium with peripheral palisading and central stellate reticulum, as well as nests of keratotic and calcifying cells (“wet” keratin) (Figure 15.8). These combined features can also be seen in odontogenic ghost cell tumors and, to a lesser extent, in calcifying odontogenic cysts (Gorlin’s cysts). Whenever feasible, complete conservative resection is the treatment of choice; adjuvant radiotherapy may be required in cases of partial excision. Prognosis worsens with age.
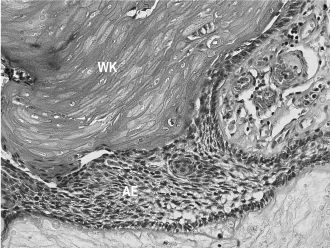
Figure 15.8. Craniopharyngioma (hematoxylin-eosin, 403). Ameloblastic epithelium (AE) with “wet” keratin (WK).
15.1.12 Ewing’s sarcoma/primitive neuroectodermal tumor
Ewing’s sarcoma/primitive neuroectodermal tumor (ES/PNET) are high-grade primitive malignancies generally occurring in bone but occasionally arising in extraskeletal sites such as the sinonasal tract. The typical onset is usually in the first 2 decades of life. Histologically, they are composed of a diffuse population of small round blue cells often containing intracytoplasmic glycogen. PNET shows neuroepithelial differentiation in the form of rosettes and pseudorosettes, and reacts to neural/neuroendocrine markers. In opposition, ES lacks neuroepithelial differentiation and neural/neuroendocrine immunoreactivity. Treatment of ES/PNET generally consists of chemotherapy and radiotherapy, although surgical management is indicated in localized disease.
15.1.13 Fibrous dysplasia
Fibrous dysplasia is considered a benign genetic fibro-osseous condition resulting in bone alteration and expansion. This rare disease can be localized to one bone (“monostotic”) or may involve multiple bones (“polyostotic”), in which case endocrine and cutaneous abnormalities may be present (McCune–Albright syndrome) as well as marked facial deformity. Fibrous dysplasia can be a component of Mazabraud syndrome, which includes intramuscular myxomas and fibrous dysplasia. The typical radiographic feature is replacement of medullary bone by an ill-defined, expansive, “ground-glass” radiopacity. SBC may be also present. Microscopic features include irregular trabeculae of woven bone lacking osteoblastic rimming and set in a cellular fibrous stroma (Figure 15.9). Treatment usually consists of bone recontouring for cosmetic purposes or to prevent compression of vital structures, although this inevitably leads to recurrence since the bulk of the lesion is not removed. Smaller lesions can be resected. Malignant transformation occurs only rarely.
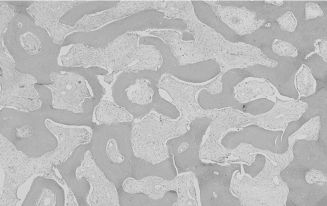
Figure 15.9. Fibrous dysplasia (hematoxylin-eosin, 103). Smooth-contoured islands of woven bone lacking osteoblastic rimming and set in a fibrous stroma.
15.1.14 Glomangiopericytoma
Glomangiopericytoma, also known as sinonasal-type hemangiopericytoma, is a rare neoplasm occurring mostly in the sinonasal tract and paranasal sinuses. Even though the very existence of hemangiopericytoma as a distinct entity is debatable, glomangiopericytoma deserves to be classified separately. It is characterized by a dense proliferation of spindled or ovoid cells arranged in a fascicular, storiform, or whorled pattern. The stroma contains abundant vascular channels, often exhibiting characteristic perivascular hyalinization. Treatment consists of complete excision. Recurrences are relatively common, even decades after surgery, and tumors may occasionally behave as low-grade malignancies.
15.1.15 Gorham’s disease
Gorham’s disease, also known as massive osteolysis or vanishing bone disease, is a very rare disease of unknown origin characterized by vascular proliferation leading to dramatic osteolysis. Patients present with spontaneous and progressive bone resorption. There is no proven cure, although successful disease stabilization through radiation therapy or i.v. bisphosphonates has been reported. Surgery alone has had unpredictable results.
15.1.16 Granular cell tumor
Granular cell tumors are relatively common benign tumors with a predilection for the tongue and larynx. They often present as asymptomatic, slow-growing, submucosal masses exhibiting a faint yellow-pink color. Laryngeal dysfunction may occur. The exact pathogenesis of this entity is unknown, although the tumor cells share some similarities with Schwann cells. Histologically, granular cell tumors are composed of sheets of large cells with abundant cytoplasm containing eosinophilic granules. A significant proportion of these tumors demonstrate marked pseudoepitheliomatous hyperplasia of the surface epithelium, which may be mistaken for an invasive squamous cell carcinoma by the untrained eye (Figure 15.10). Conservative excision is the treatment of choice.
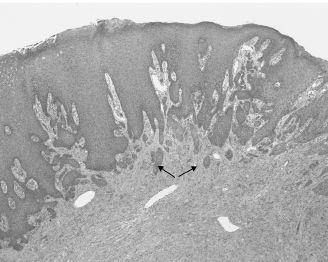
Figure 15.10. Granular cell tumor. The surface epithelium displays prominent pseudoepitheliomatous hyperplasia (arrows), mimicking superficially invasive squamous cell carcinoma (hematoxylin-eosin, 103).
15.1.17 Langerhans cell histiocytosis
Langerhans cell histiocytosis is characterized by a proliferation of Langerhans cells that can either be localized (eosinophilic granuloma) or multifocal with possible multiorgan involvement. In the head and neck area, it typically presents as an intraosseous lesion. Langerhans cells are large antigen-presenting cells with grooved or convoluted nuclei. Numerous eosinophils are typically seen admixed with the tumor cells (Figure 15.11). The ultrastructural hallmark of Langerhans cells is the Birbeck granule, a disk- or tennis racquet-shaped cytoplasmic organelle. Eosinophilic granulomas respond well to conservative surgery, whereas multifocal forms require chemotherapy. Malignant versions of Langerhans cell histiocytosis have been described.
15.1.18 Leiomyoma
Leiomyomas are benign tumors of smooth muscle that typically present as well-defined, occasionally painful, submucosal masses. Noncutaneous leiomyomas can arise in the walls of vascular channels (vascular leiomyomas or angioleiomyomas). Histologically, they are composed of fascicles of mature spindle cells with blunt-ended nuclei and perinuclear vacuolization. Different histologic variants have been described, such as the solid, granular cell and epithelioid types. Conservative excision is usually curative.
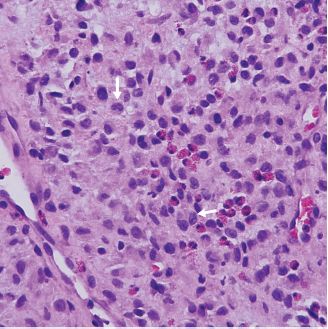
Figure 15.11. Langerhans cell histiocytosis. Scattered Langerhans cells with pale cytoplasm and grooved or convoluted nuclei (arrows). Eosinophils are also present (hematoxylin-eosin).
15.1.19 Lipoma
Lipomas are benign tumors of adipose tissue and represent the most common benign mesenchymal tumors. Distinct histologic variants have been described, such as pleomorphic, myxoid, chondroid, spindle-cell, intramuscular (infiltrating), and so forth. Complete surgical excision is the treatment of choice in many, if not all, types of lipomas. Recurrence is rare.
15.1.20 Liposarcoma
Liposarcomas are among the most common soft tissue sarcomas. In the head and neck, they occur most commonly in the larynx, hypopharynx, and neck. Much like their benign counterparts, liposarcomas are classified into numerous subtypes. The histologic hallmark of liposarcomas is the presence of vacuolated lipoblasts, which can be frustratingly lacking in well-differentiated tumors. Treatment and prognosis depend on histologic variants. Well-differentiated and myxoid variants have the best 5-year survival rate.
15.1.21 Lymphangioma
Lymphangiomas are benign lesions of lymphatic channels. They have a predilection for the head and neck and can be seen in the setting of genetic or congenital anomalies. Lymphangiomas are sometimes classified into three histologic types: cystic (cystic hygroma), capillary, and cavernous. Cystic hygromas are most often present at birth and may undergo spontaneous involution during childhood. They typically spread along tissue planes and may extend downward into the thoracic cavity. Cystic hygromas are characterized by dilated lymphatic spaces containing proteinaceous fluid. The treatment of choice is complete surgical excision, although sclerotherapy and laser therapy have been useful in selected cases. Even though lymphangiomas remain benign, death may occur as a result of pulmonary complications or impingement on vital structures.
15.1.22 Malignant peripheral nerve sheath tumors
In the head and neck, malignant peripheral nerve sheath tumors (MPNST) are rare and have a predilection for the neck. Although they may arise de novo in peripheral nerves, they usually develop in the setting of NF type 1 or from a preexisting BPNST. Histologically, MPNST are often composed of spindle cells arranged in long intersecting fascicles. Heterologous elements, such as metaplastic bone and cartilage, can be seen. MPNST can also exhibit divergent differentiation (muscular or glandular), in which case synovial sarcoma and melanoma should be ruled out. Low-grade tumors contain very few atypical mitotic figures and no necrosis, resembling atypical BPNST; therefore, demonstration of infiltrative growth is essential. Treatment includes wide resection with postoperative radiotherapy and chemotherapy. Prognosis is poorer in NF-1-related tumors.
15.1.23 Meningioma
Meningiomas are common benign intracranial neoplasms of meningothelial cells, although extracranial tumors are possible. Meningiomas are usually slow-growing, and may cause bone erosion and extension into the central nervous system. Different histologic subtypes have been described. Among these, the meningothelial subtype is the most common in extracranial sites of the head and neck. It is characterized by the presence of whorls and confluent islands of epithelioid cells with occasional nuclear inclusions (Figure 15.12). Concentric calcifications (psammoma bodies) can be seen but are not required for the diagnosis. Complete resection is the treatment of choice; recurrences are common in cases where surgical access is limited.
15.1.24 Nodular fasciitis
Nodular fasciitis is a benign, fast-growing, tumor-like proliferation of myofibroblasts that is part of the same spectrum of disease as cranial
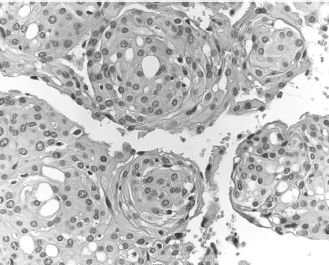
Figure 15.12. Meningioma. Whorls of epithelioid tumor cells (hematoxylin-eosin).
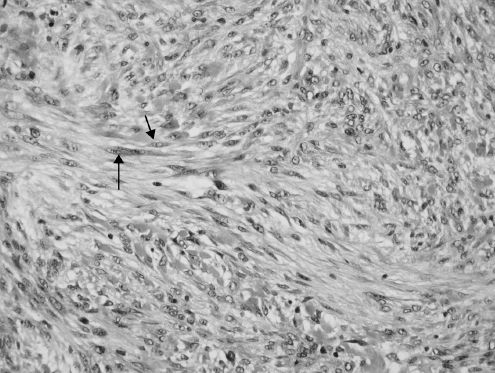
Figure 15.13. Nodular fasciitis. Fascicles of bipolar spindle cells (arrows) (hematoxylin-eosin).
fasciitis and intravascular fasciitis. It may affect any age, although young patients are more often affected. Trauma is thought to be a possible etiologic factor. Histologically, nodular fasciitis is vaguely infiltrative and composed of fascicles of immature bipolar spindle cells that can display a fair amount of pleomorphism and typical mitotic figures (Figure 15.13). The stroma contains inflammatory cells and extravasated red blood cells, and also exhibits keloidal collagen deposition as the lesions age. Misdiagnosis as a sarcoma is a common diagnostic pitfall. Conservative surgical excision is generally considered curative.
15.1.25 Osteoblastoma
Osteoblastomas are benign bone-forming tumors. By definition, the term “osteoid osteoma” is used instead when the lesion is less than 2 cm in its greatest dimension. Osteoblastomas can occur in any bone but have a predilection for the mandible. The male-to-female ratio is roughly 2:1, and patients in the second decade are most often affected. Nocturnal pain is a characteristic symptom. Histologic features include anastomotic trabeculae of woven bone and osteoid with prominent osteoblastic rimming set in a markedly vascularized intertrabecular stroma. Osteoblastomas are analogous to cementoblastomas (not discussed in this chapter) except that the latter are attached to dental roots. Complete excision is the treatment of choice.
15.1.26 Osteoma
Osteomas are benign slow-growing tumors of the craniofacial bones (Figure 15.14). Multiple osteomas of the maxillofacial complex are pathognomonic of Gardner’s syndrome (gastrointestinal adenomatous polyps, osteomas, skin cysts, and numerous types of tumors). Osteomas are to be differentiated from exostoses and tori, which are hamartomatous by nature and lack significant growth potential. The histologic appearance of osteomas is one of mature and predominantly lamellar bone. Nonsyndromic osteomas are cured by local excision, although surgery is recommended only in the presence of symptoms or functional disturbance.
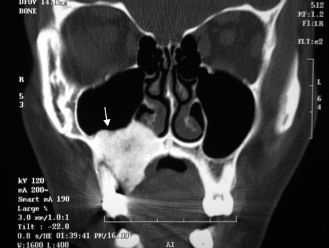
Figure 15.14. Osteoma. Coronal computed tomogram showing a dense mass of the right maxillary sinus (arrow).
15.1.27 Osteoradionecrosis
Osteoradionecrosis (ORN) is a relatively common complication of radiation treatment with a predilection for the mandible. Irradiated bone becomes less vascularized, hypoxic, and hypocellular, thus less able to regenerate or respond to infections. ORN is rare in patients who received radiation doses less than 5000 cGy. Acute and chronic infections often complicate ORN. Early cases may not be visible through imaging studies. Chronic ORN may present just like chronic osteomyelitis, i.e., as ill-defined, mixed radiolucent-radiopaque lesions with periosteal duplication, formation of sequestra, and possible pathologic fracture (Figure 15.15). Histologic examination reveals bone trabeculae deprived of osteocytic lacunae set in inflamed fibrous tissue. Epithelialized fistulous tracts with reactive cytologic atypia may be seen. Biopsy specimens of ORN must be thoroughly inspected to rule out radiation-induced sarcomas or recurrent disease. Treatment ranges from conservative curettage to resection of diseased bone, with cessation of smoking. Most studies indicate that hyperbaric oxygen therapy is a useful therapeutic adjunct.
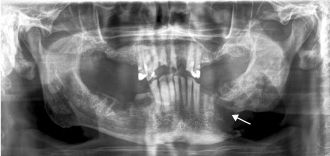
Figure 15.15. ORN. Panoramic radiograph showing an irregular pathologic fracture of the left mandibular body (arrow).
15.1.28 Osteosarcoma
Osteosarcoma is the most common primary malignancy of bone. It can be classified according to its localization, whether intraosseous (conventional), juxtacortical, orextraosseous, and according to the histologic grade. Regardless of the histologic variant, all osteosarcomas demonstrate production of either malignant bone or osteoid (Figure 15.16). Differentiating low-grade osteosarcomas from osteoblastomas or even fibrous dysplasia can be an arduous task on small biopsies; therefore, clinicopathologic correlation is paramount. Distinguishing osteosarcomas from giant cell tumors can also prove to be challenging in lesions with little osteoid and numerous giant cells. Treatment consists of wide resection with tumor-free margins, possibly with neoadjuvant or postoperative chemotherapy.
Osteosarcomas of the jaw have a distinctly better prognosis than extragnathic tumors. Nevertheless, tumors arising in irradiated bone or in preexisting bone lesions, such as Paget’s disease of bone, tend to have a poorer prognosis.
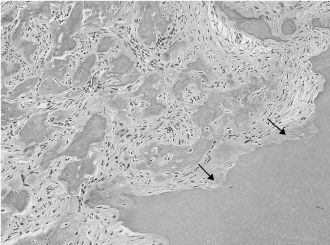
Figure 15.16. Well-differentiated osteosarcoma (hematoxylin-eosin). Somewhat atypical stromal cells associated with the formation of osteoid and bone. Adjacent dentin is showing irregular resorption (arrows).
15.1.29 Paraganglioma
Paragangliomas are slow-growing neuroendocrine neoplasms arising from paraganglia, some of which are distributed in the jugular bulb (glomus jugulare), the medial cochlea promontory (glomus tympanicum), the carotid, and the vagal body. They are morphologically similar to pheochromocytomas but are not to be confused with glomus tumors or glomangiomas, which are entirely different entities and are exceedingly rare in the head and neck. A significant proportion of paragangliomas is linked to inherited mutations (hereditary paragangliomatosis) and must be ruled out in multicentric tumors. Histologically, these tumors are composed of nests (“Zellballen”) of granular tumor cells surrounded by spindled sustentacular cells (Figure 15.17
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


