Fig. 3.1
Direct cell counting. Cells growing on two different substrata (a vs. b) were loaded with DCFH-DA for 24 h, and fixed with neutral formalin. More cells are to be counted in (b)
The indirect methods for the estimation of the cell number are mostly based on the absorption of dyes or fluorescent molecules in the cells, thus allowing comparisons of the cell numbers in the treated vs. the control cultures, by spectrophotometric or spectrofluorimetric techniques. Most of the dyes used are absorbed in the cells’ proteins or in the cellular nucleic acids, and then they are solubilized by an appropriate solvent, so as to enable the spectrophotometric estimation of the amount of the absorbed dye. The most common dyes are the following: methylene blue [26–29], crystal violet [3, 30, 31], Coomassie blue [32], sulphorhodamine B [33], neutral red [24, 34–37], BCECF [2′-7′-biscarboxyethyl-5(6)-carboxyfluorescein] [38], PI [propidium iodide] [39, 40], Hoechst 33258 [41–43], alamar blue [44] and fluorescein diacetate [45]. Some of these, such as neutral red, BCECF or fluorescein diacetate, stain only living cells. They are called vital dyes, and they can be used to determine the viability of the cells, i.e., the percentage of living cells in the total cell culture. But the most widely used method for assessing viable cell numbers is the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay [46], which is based on the reduction of this dye by the mitochondrial enzymes to its formazan product and the concomitant change in its absorption spectrum (Fig. 3.2). Many modifications and re-evaluations of the method have appeared [3, 16, 36, 47–50], and moreover, other tetrazolium salts have been developed, which can replace MTT [51–53].
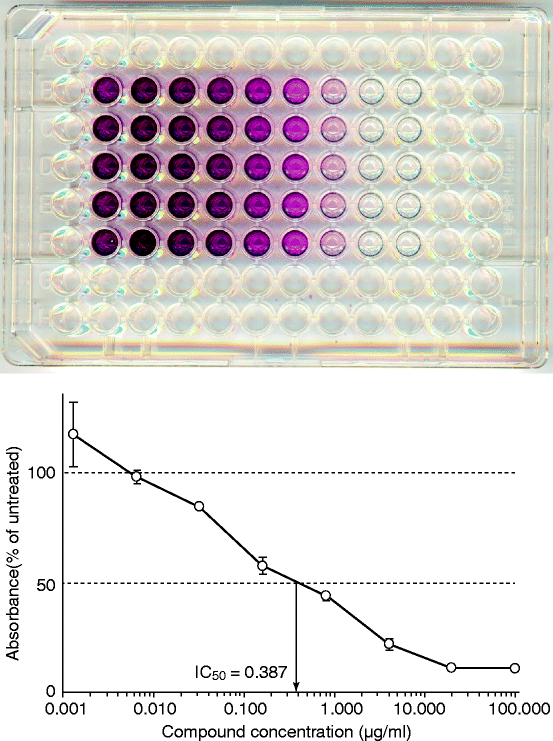
Fig. 3.2
Indirect assessment of viable cell number by the MTT method. A representative 96-well plate is shown after treatment of the cells with a cytotoxic agent for 48 h and the formation of MTT-formazan by the living cells (upper panel). After counting the absorbance of each well in a microplate reader, the concentration of the agent that eliminates 50 % of the cells (IC50) can be estimated (lower panel)
Another way of indirect estimation of the cell number is by following the uptake of radiolabeled molecules necessary for the cell functions, such as amino acids [54, 55] or nucleotides, using either autoradiography or liquid scintillation counting [16, 56, 57]. Again these methods are dependent on the viability of the cell culture, since the radiolabeled precursors have to be metabolically incorporated in the cells, and not passively absorbed. Especially regarding incorporation of [3H]-thymidine into newly synthesized DNA, there has been some criticism, since (1) changes in [3H]-thymidine incorporation may relate to changes in the intracellular nucleotide pools or in the activities of key enzymes, such as thymidine kinase, rather than to changes in DNA synthesis; and (2) there are drugs, like 5-fluorouracil or methotrexate, which cause increased uptake of [3H]-thymidine through the salvage pathway, although they inhibit DNA synthesis [58]. In spite of these drawbacks, this is perhaps the most widely used method for assessing the proliferative index of a culture. Furthermore, the method can be used also for the determination of the distribution of the cells in the various phases of the cell cycle, so it provides some clues on the mechanism of action of the test compound.
Nowadays, there is a tendency to replace radioactivity-based techniques with nonradioactive ones, since the former require specific set-up and licence, not available in all laboratories, and they pose potential risks for the personnel. In this vein, an alternative to [3H]-thymidine incorporation for the estimation of DNA synthesis is the incorporation of the thymidine analog BrdU (5-bromodeoxyuridine) and its subsequent tracing in situ with the help of a specific antibody [59–61]. As shown in Fig. 3.3, the nuclei of the cells that progress through S phase incorporate BrdU and can be visualized by immunofluorescence. Although this method is much more reliable than [3H]-thymidine incorporation for the estimation of the percentage of cells in the S-phase of the cell cycle, it is more laborious and, consequently, inappropriate for the screening of large amounts of substances.
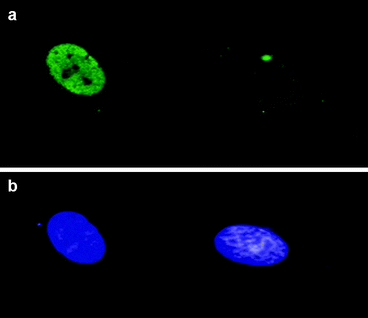
Fig. 3.3
Evaluation of BrdU incorporation by immunofluorescence. The nucleus of a cycling cell (left) incorporated BrdU in contrast to an arrested one (right), as shown using a monoclonal antibody against BrdU and FITC-conjugated anti-mouse-IgG (panel a). In panel b, the same nuclei were counterstained with DAPI (magnification 1,000×)
Here, one should mention separately the flow cytometric techniques, which give fast and accurate results from large cell populations. Staining of the cells to be used for flow cytometry is accomplished as already mentioned above; however, the information obtained is at the single cell level and multi-parametric. So after staining of the DNA with propidium iodide (see above), the DNA content of each cell from a given cell population can be estimated, thus allowing determination of the distribution of the cells in the various phases of the cell cycle [62–66]. As an example, typical histograms of PI-fluorescence produced by a FACScalibur flow cytometer (Becton Dickinson) using the ModFit software (Verity) are shown in Fig. 3.4, where the left peak corresponds to the number of cells at G0/G1 phase and the right one to those at G2/M. Using fluorescent antibodies and flow cytometry, the percentage of cells that are synthesizing DNA can also be estimated after BrdU incorporation (see above) [67]. Furthermore, the simultaneous detection of the expression of certain proteins, together with the above markers can give information on the mechanism of action of the test compound [68]. However, the high cost of flow cytometric techniques and the requirement of processing each sample individually are limiting their use for cases where relatively small sample numbers are being studied.
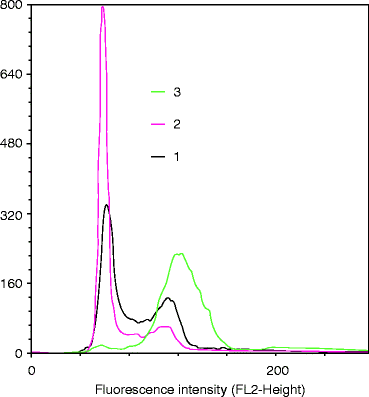
Fig. 3.4
Flow cytometric cell-cycle analysis using propidium iodide. The distribution of human cells in the various phases of cell cycle was assessed by flow cytometry after PI staining (1 untreated cycling culture, 2 culture arrested at the G0/G1 phase, 3 culture arrested at the G2/M phase)
3.3.2 Cell Death Assays
Direct toxicity represents a major problem for the use of several biomaterials. For the determination of cell death there are stains special for dead cells, such as trypan blue, which was traditionally used due to its exclusion by cells with undamaged cytoplasmic membranes [25, 69, 70]. Other substances used in “dye exclusion tests” are fast green [71], erythrosin B [72], and dansyl-lysine [73]. On the other hand, one can assess the cytotoxicity of the materials under examination by estimating the percentage of the cells in the culture which are living, using the above mentioned vital stains, e.g., neutral red, Hoechst 33258, BCECF or fluorescein diacetate, always in comparison to a control culture.
In another approach, cells are preloaded with a radioactive compound and then assayed for leakage of radioactivity from the cells with damaged membranes. Traditionally, [51Cr] has been used for such studies [74–77], but nowadays the trend is to substitute for it with the nonradioactive method of europium release [78]. A disadvantage of both the dye exclusion and the radioactive compound release methods is the underestimation of reproductively dead cells, i.e., those incapable of proliferation [78, 79]. A more simple method to obtain quantitative information about the cells with damaged cytoplasmic membranes is the spectrophotometric measurement of the released lactic acid dehydrogenase (LDH) in the culture supernatant [80].
Since there are two types of cell death – necrosis and apoptosis – it is important in several cases to identify which type of cell death is occurring in order to understand the mechanism of the cytotoxic action of a compound. Apoptosis or programmed cell death follows a predetermined series of events, during which the chromatin becomes fragmented and condensed, the organelles and the cell shrinks, and the cell surface blebs, leading to budding off of membrane-bound apoptotic bodies [81]. These morphological alterations allow monitoring of apoptosis in several ways. One common approach is to document the appearance of DNA laddering by gel electrophoresis [80, 82]. Since this method is not applicable in all cell types [83] and does not allow analysis of a large number of samples, the staining with nucleic acid fluorochromes, such as propidium iodide or DAPI (4,6-diamidino-2-phenylindole) is preferred, followed by microscopic inspection for the assessment of DNA fragmentation [84] (see Fig. 3.5a–b). Actually, this method can become quantitative only in combination with flow cytometry, so that one can determine the percentage of the cells with reduced DNA content, i.e., hypodiploid cells [63, 85] (Fig. 3.5c–d). Furthermore, DNA fragmentation can be visualized after addition of labeled nucleotides in the DNA breaks in a reaction catalyzed by exogenous terminal deoxynucleotidyl transferase (TUNEL method) either in situ by fluorescence microscopy or by flow cytometry [80, 86, 87]. On the other hand, alterations in membrane integrity which occur much earlier than DNA fragmentation in apoptotic cells can be detected after staining with annexin V [88, 89] or the viable dye merocyanine 540 [90]. Another early event in apoptosis is loss of the mitochondrial inner transmembrane potential [91, 92]. This can be monitored by using rhodamine 123 [93, 94], 3,3′-dihexylocarbocyanine iodide (DiOC6) or 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) [95], as well as MitoTracker Green FMTM or RedTM CMXRos [96] in conjunction with flow cytometry or confocal microscopy.
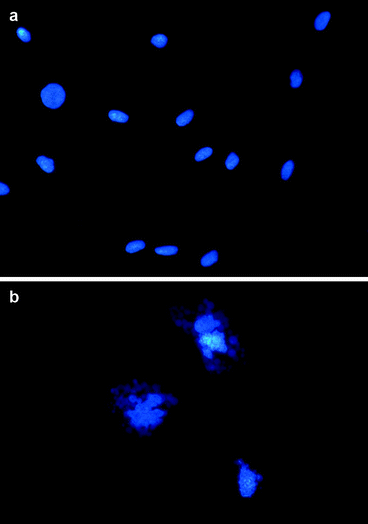
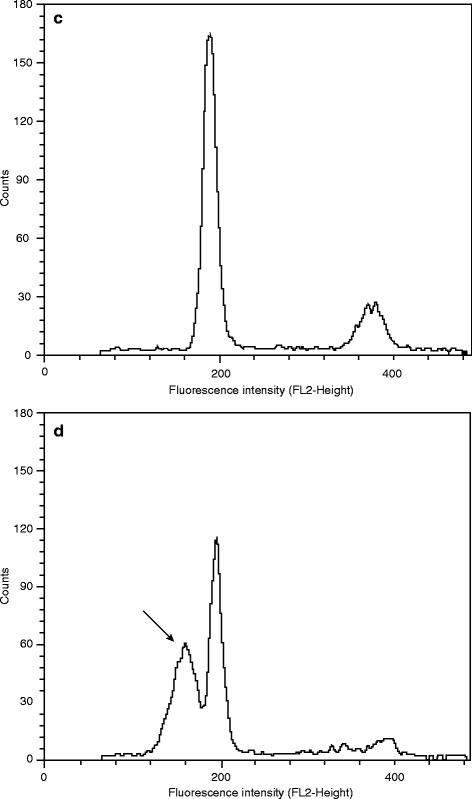
Fig. 3.5
Detection of apoptosis. The nuclei of control (a) and apoptotic (b) cells were stained with DAPI after fixation with neutral formalin and permeabilization with Triton X-100 (magnification 1,000×). Alternatively, the distribution of human cells in the various phases of cell cycle was assessed by flow cytometry as in Fig. 3.4 (c: control culture, d: culture undergoing apoptosis). The arrow indicates the sub-diploid peak due to apoptosis
3.4 Other Assays
A common mechanism for the induction of cell death by several materials used in the dental practice is through oxidative stress [97]. Accordingly, many studies of the cytotoxicity of certain materials include their ability to act as oxidants and to promote the production of reactive oxygen species (ROS) inside the cell. Often the oxidative properties of the materials are initially evaluated in cell-free systems, e.g., by their interaction with the free radical diphenylpicrylhydrazyl (DPPH), followed by spectrophotometric monitoring [98, 99]. A more novel, cell-based technique is the evaluation of the material’s ability to induce oxidative stress inside the cell using a convenient fluorochrome, i.e., 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), a cell-permeable nonfluorescent probe, which is de-esterified intracellularly and turns to the highly fluorescent 2′,7′-dichlorofluorescin upon oxidation [98, 100–103]. Further evaluation of the oxidative status of the target cells may include monitoring of intracellular glutathione depletion [97, 104, 105] and glutathione transferase activity [106], but these methods are laborious and time-consuming, not allowing for the screening of extensive numbers of materials. Another parameter that needs to be tested in biomaterials under study is their genotoxic potential, i.e., their ability to provoke DNA damage. Single cell gel electrophoresis (comet assay) followed by labeling of the samples with a fluorophore such as DAPI can reveal chromatin fragmentation in the form of single- or double-strand DNA breaks [107, 108]. When visualized under a fluorescent microscope, damaged DNA appears as a comet tail that trails the comet head containing the undamaged nuclear DNA (Fig. 3.6).
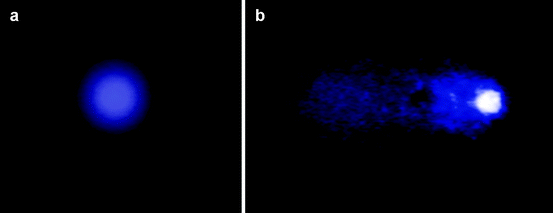
Fig. 3.6
Assessment of DNA integrity by single cell gel electrophoresis. Cells were detached, electrophoresed in low-melting point agarose and stained with DAPI. (a) Nucleus containing intact DNA; (b) nucleus with damaged DNA. Damaged DNA appears in the “tail” of the “comet”
In general, after the evaluation of the possible cytotoxic, genotoxic and/or antiproliferative activities of the test materials, more specific targets are chosen, usually to investigate the effects on certain cellular products. However, a detailed presentation of such methods is beyond the scope and the space limitations of the this chapter. Briefly, assays monitoring gene and protein expression are used, such as RT-PCR [109] or northern blotting [110] and western blotting, ELISA [111], immunofluorescence [60, 112] or flow cytometry [113, 114], respectively. Phosphorylation of specific intracellular substrates [115] or activity of certain transcription factors [21, 116] can also provide important information regarding the intracellular signaling mechanisms affected by the test compounds. Interestingly, novel technologies such as the use of DNA microarrays for gene expression profiling have also been added to the toolbox of the researchers nowadays [117, 118].
3.5 Important Parameters
In all of the in vitro assays described above, there are a number of common parameters, which are critical for the outcome of the assay. Perhaps the most important are the concentration spectrum of the evaluated compound, as well as the time of exposure. Usually, the concentrations used in the assay are dictated by the background knowledge on similar compounds and also by the application to which the test compound is aiming. Of course, the concentration range chosen should always result in a dose–response curve. It should be mentioned here that the term “concentration” is applicable only for soluble compounds; for certain solid materials, a different approach of testing is followed by growing the cell cultures in direct contact with the material [119–121], although such systems are suitable mostly for qualitative studies (see also alternative culture systems below).
For the choice of the exposure time, one should know exactly the duration of the cell cycle in the cultures used for the assay. Usually, exposure for a period of one to two cell cycles is enough for the manifestation of the proliferative, cytostatic, or cytotoxic action. However, there are cases where longer exposure times could be required, since prolonged incubation with a variety of cancer chemotherapeutic agents has been shown to result in gradually decreasing IC50 values, as exposure time increases [55]. Undoubtedly, only a prolonged exposure will demonstrate whether a cell population remains clearly unaffected by the test compound. Especially for materials used in dental practice, the study of the possible slow release of putatively harmful agents due to material aging or corrosion requires the examination of lower concentrations for longer incubation times, thus simulating more closely the in vivo conditions. Furthermore, in some instances a recovery period after exposure to the test compound is included in the assay, either to allow for the occurrence of delayed effects or to permit the recovery of metabolic perturbations which could affect the index of the assay per se.
Other important issues may occur mainly from the nature of the test materials; for example, false positive results have been reported to arise due to release of unexpectedly volatile compounds from one extract affecting the neighboring cultures [122]. Precise knowledge of the physicochemical properties of the test materials is crucial for the design of each assay, as well as to avoid the wrong interpretation of the results obtained.
3.6 Alternative Culture Systems
While in conventional assay systems cells are cultured on plastic surfaces, there is now the trend to create three-dimensional cultures using extracellular matrix components, such as collagen gels [123, 124] or fibronectin-coated meshes [125–127]. In such more complex environments, one can take into account the mutual and organizational interactions between cells and their surrounding matrix, under conditions more closely simulating the tissue milieu [123]. In analogous approaches, it has been proposed to test orthodontic materials on reconstituted human oral epithelium produced by keratinocytes cultured on inert polycarbonate filters [128, 129], as well as dental restorative materials in the presence of a dentine barrier, since the interaction with the latter may affect the cytotoxicity of the materials [130, 131].
In conclusion, biocompatibility testing is fundamental for the development of novel biomaterials. In this chapter, we have presented a plethora of in vitro assays of vital cellular functions, such as cell proliferation or cell survival, which can provide a first clue for the biocompatibility of a given material. Innovative methods are continuously being developed, as well as improvements of the existing ones, although the basic principles of all of these assays remain generally the same as the ones presented here.
References
1.
Williams DF (1987) Definitions in biomaterials. Proceedings of a consensus conference of the European society for biomaterials, Chester, England, March 3–5, 1986, 4. Elsevier Science Ltd, New York
2.
Wataha JC (2001) Principles of biocompatibility for dental practitioners. J Prosthet Dent 86(2):203–209PubMed
3.
Bester MJ, Erasmus F, Pretorius E (2003) Presenting an in vitro cell culture model to determine the cytotoxic effect of benzoyl peroxide vapours. SADJ 58(5):183–186, 188PubMed
4.
Hauman CH, Love RM (2003) Biocompatibility of dental materials used in contemporary endodontic therapy: a review. Part 1. Intracanal drugs and substances. Int Endod J 36(2):75–85PubMed
5.
Autian J (1970) The use of rabbit implants and tissue culture tests for the evaluation of dental materials. Int Dent J 20(3):481–490PubMed
6.
ISO (1984) Biological evaluation of dental materials. International Organization for Standardization Technical Report:7405
7.
Al-Hiyasat AS, Darmani H (2005) The effects of recasting on the cytotoxicity of base metal alloys. J Prosthet Dent 93(2):158–163PubMed
8.
Nelson SK, Wataha JC, Lockwood PE (1999) Accelerated toxicity testing of casting alloys and reduction of intraoral release of elements. J Prosthet Dent 81(6):715–720PubMed
9.
Watanabe I, Wataha JC, Lockwood PE, Shimizu H, Cai Z, Okabe T (2004) Cytotoxicity of commercial and novel binary titanium alloys with and without a surface-reaction layer. J Oral Rehabil 31(2):185–189PubMed
10.
Mockers O, Deroze D, Camps J (2002) Cytotoxicity of orthodontic bands, brackets and archwires in vitro. Dent Mater 18(4):311–317PubMed
11.
Wataha JC, Hanks CT, Sun Z (1994) Effect of cell line on in vitro metal ion cytotoxicity. Dent Mater 10(3):156–161PubMed
12.
ISO (1999) Biological evaluation of medical devices – part 5: tests for in vitro cytotoxicity. International Organization for Standardization 10993-5:1999(E), 2nd edn
13.
Adams RLP (1980) Cell culture for biochemists. Elsevier, Amsterdam, pp 181–201
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


