Fig. 8.1
Hematoxylin/eosin staining (left panels) and polarized light (right panels) micrographs of cortical bone of mouse proximal tibia (original magnification ×4)
Cells of mononuclear origin express the band 5 isoenzymes of tartrate-resistant acid phosphatase (TRAP) [12, 13]. This enzyme is characterized by cathodal electrophoretic mobility at pH 4 and by resistance to inhibition by L(+)-tartate [14]. In mammals, TRAP has been detected in several tissue systems as a minor acid phosphatase isoenzyme [15, 16]. Nonetheless, it is primarily expressed in bone-resorbing multinucleated osteoclasts of the skeleton [12, 17]. The function of TRAP remains obscure. Several studies have proposed that in resorbing osteoclasts, TRAP is localized in the ruffled border or in the Howship lacunae [18]. However, in vitro and immunoelectron microscopy studies have documented that TRAP is also located in large transcytotic vesicles [19, 20]. Therefore, degradation of extracellular matrix proteins (namely, bone sialoproteins, osteopontin, and osteonectin) occurs in both the Howship lacunae and the intracellular transcytotic vesicles [21]. TRAP can function as an excellent, highly specific osteoclast marker, which can be easily detected by commercially available histochemical kits. The histochemical TRAP staining results can be evaluated under light microscopy. The TRAP-positive osteoclast surface and the TRAP-positive osteoclast number can be calculated either manually or with the use of proper software [22, 23]. This stain can be applied on both decalcified and non-decalcified tissues [12, 17, 23]. The development of more sophisticated TRAP protocols, such as fluorescent-based TRAP stains, holds promise for better visual results and can be combined with other immunofluorescent, as well as immunohistochemical methods [24]. Example of TRAP labeling is shown in Figs. 8.2 and 8.3.


Fig. 8.2
Tartrate-resistant acid phosphatase (TRAP) labeling of mouse femur. Note densely colored areas that represent bone-resorbing osteoclasts (original magnification ×4)
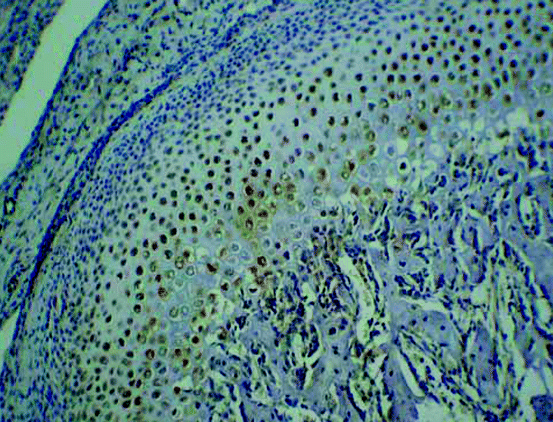
Fig. 8.3
Higher-magnification micrograph of area shown in Fig. 8.1, highlighting TRAP-positive osteoclasts (original magnification ×20)
A commonly used marker of osteogenic development is alkaline phosphatase, an enzyme that was discovered by Robison in 1923 [25]. The term ALP was subsequently introduced in 1979 by McComb and colleagues [26]. Four distinct genes that encode for 4 different ALP isoenzymes have been discovered in humans: intestinal, placental germ-like, and tissue-non-specific (TNS). TNSALP is expressed in liver, bone, and kidney [27, 28]. The biological role of this protein is largely unknown. Within bone tissue and teeth, ALP is produced by osteoblasts and is involved in the process of osteoid mineralization [29]. This function is facilitated by local elevation of inorganic phosphate and destruction of inhibitors of hydroxyapatite crystal growth, phosphate transportation, and ATPase or tyrosine-specific phosphoprotein phosphatase activity [30]. Numerous studies of bone and cartilage development have highlighted the importance of measurement of ALP for the evaluation of osteoblastic activity. The identification of ALP activity in tissue sections is made primarily by histochemical methods [31, 32]. These methods are applied mainly to non-decalcified, frozen bone, and cartilage tissues. Indeed, ALP is sensitive to decalcification procedures since they remove the zinc and magnesium ions that are essential for ALP activity [31]. ALP is expressed in stimulated osteoblasts, bone-lining cells, and some newly formed osteocytes as well as in pre-apoptotic chondroblasts. Recently, histochemical methods that can be applied to decalcified, paraffin-embedded skeletal tissue have been developed [33, 34]. These methods are relatively easy, cheap, and reproducible and can be used in conventional pathology/histology laboratories.
In addition to histochemisty, immunohistochemical methods have been developed for the detection of ALP activity and localization [32, 35], using polyclonal antibodies against TNSAP or tissue-specific monoclonal antibodies against the bone isoform [32, 36, 37]. Histochemical and immunohistochemical approaches provide an in situ estimation of ALP localization and function, as they reveal differential localization of the examined enzyme during the different steps of bone and cartilage development and maturation.
Another commonly used assay for the study of bone maturation and mineralization was described in 1901 by von Kossa [38]. An example of this assay is shown in Fig. 8.4.
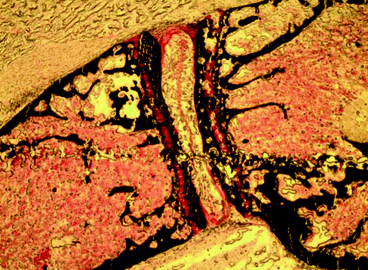
Fig. 8.4
Von Kossa assay: micrograph of mouse femur. Black areas represent sites of calcium deposition, whereas red areas correspond to osteoid (non-calcified bone) and collagen (original magnification ×4)
The von Kossa assay is an excellent method for the detection of calcium depositions. Notably, this stain does not react with calcium but with phosphate and carbonate ions in the presence of acid material [39]. More specifically, this method is based upon the principle that cationic silver ions can be removed from solution by carbonate or phosphate ions because of their respective positions in the electrochemical series. When undecalcified tissue sections are treated with 5 % silver nitrate solution, cationic silver replaces calcium in the original salt and forms a silver salt that can be displayed by a reduction to metallic silver. This reaction is photochemical, and the activation energy is supplied from strong violet or ultraviolet light [40, 41]. Silver ions are associated with phosphate ions, and therefore they are considered to indirectly uncover calcium deposits. After the treatment with silver nitrate and aqueous sodium thiosulfate, counterstaining is required. For this purpose, van Gieson and hematoxylin-eosin stains are recommended. It can be seen in Fig. 8.4 that when the von Kossa assay is completed, sites of calcium deposition are stained black, and the osteoid and collagen are stained red, whereas fibrous tissue and red blood cells are stained yellow.
In addition, the H&E stain highlights the cellular components of the examined sections, providing important information regarding the histology of the examined tissues. The von Kossa assay is a very simple, accurate, and inexpensive technique for the study of bone maturation and mineralization. Furthermore, von Kossa-stained sections can be used for histomorphometric analyses. The purpose of histomorphometry is the evaluation of the structural integrity of the skeleton, the degree of bone formation and mineralization, and the rate of bone resorption. The tested parameters that reflect skeleton structural integrity are the total bone volume, the volume of cancellous bone, and the amount of trabecular osteoid. The parameters that are associated with bone formation and mineralization are the surface of the trabecular osteoid, the surface of mineralization, the distance between two tetracycline-pulse labels per day (see Sect. 8.2.3), and the mineralization lag time. Finally, the factors that indicate osteoclast-resorption function are: the trabecular, cortical, and periosteal resorptive surfaces; the trabecular osteoclast count (number of osteoclasts per area); and the cortical porosity (percentage of the cortex that contains pores without osteoclasts) [42, 43]. Histomorphometry is a method of choice for the study of conditions such as metabolic bone diseases, neoplasias, bone remodeling, and fracture repair, as well as bone-cartilage response to biomechanical stress [43].
8.2.3 Fluorescent Labeling
Bone tissue continually undergoes shape and structure changes. Accretions in length and thickness, modeling, and drift activities lead to morphologic alterations that determine the relative position between various skeletal parts. Metabolic bone diseases, bone repair processes, mechanical stress, and aging create new functional demands and are responsible for the subsequent structural adaptations. An accurate method for the detection of such structural/functional modifications is fluorescent labeling [44]. Fluorescent double-labeling is used to calculate kinetic data on bone turnover. The fluorochromes are administrated systematically and form long-lasting chelate complexes with apatite, via their active iminodiacetic acid groups. Hence, they can serve as markers that allow the identification of mineralized tissues [45]. Different types of fluorochromes, such as yellow tetracyclines, xylenol orange, alizarin red derivatives, or green fluorescein derivatives like calcein (Fig. 8.5) or DCAF, which produce different colors, are available [46, 47]. The first dose of fluorescent dye is incorporated in the newly formed bone at the bone-osteoid interface, where it appears as a linear fluorescence under UV light microscopy. The second dose is administrated 3–14 days after the first. The amount of bone that has been synthesized during this time period can be calculated by measuring the width between the two lines of fluorescence. Dosing of the tetracycline is dependent upon the model individual, such as human, rat, or rabbit.
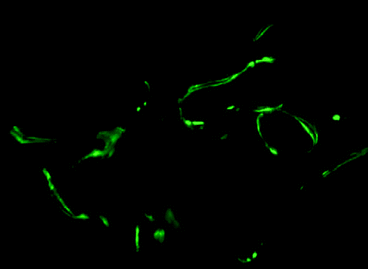
Fig. 8.5
Epifluorescence images of vertebral body from mouse following administration of two calcein labels, 3 days apart (original magnification ×10)
8.2.4 Immunohistochemistry (IHC)
Immunohistochemistry is used in everyday practice at pathology and histology laboratories. It is a relatively simple method for the in situ detection of proteins. IHC is based on the principle that specific intra- or extracellular antigens are bound to monoclonal or polyclonal antibodies that are associated with specific enzymes. The detection of the antigen-antibody complex is achieved with the use of chromogens. The principal enzyme that facilitates the antibody detection is peroxydase, and the chromogen that is most commonly used is 3,3′-diaminobenzidine tetrahydrochloride (DAB). There are two major categories of IHC methods: direct and indirect. Indirect IHC methods include peroxydase-antiperoxydase (PAP), avidin-biotin complex (ABC), and biotin-streptavidin assay (B-SA), which is the most popular [48]. The B-SA IHC method relies on the non-immunologic binding of biotin to streptavidin (a 60-kD protein), produced by Streptomyces avidinii. Three reagents are used: (a) the primary antibody, which is specific for the antigen of interest; (b) the secondary (biotinylated) antibody that binds the first one; and (c) the streptavidin-peroxidase reagent that is associated to the secondary antibody. IHC provides an in situ approach to investigate the expression and activation status of the examined proteins.
Furthermore, this is a useful method for bone biology studies since it detects the expression levels of several proteins implicated in bone development and growth. Among them, osteonectin (ON) and osteocalcin (OC) are the most significant. Examples are shown in Figs. 8.6 and 8.7. ON is a 35–45-kD protein that has the ability to bind to Ca+2, hydroxyapatite, and collagen [49]. Among its structural features, the two EF-hand high-affinity calcium-binding sites are functionally the most significant. ON mediates the deposition of hydroxyapatite and is involved in the regulation of the osteoblastic cell cycle and bone maturation. OC (also called bone gla protein) is a 5-kD protein that belongs to a family of extracellular matrix proteins named gla proteins. OC possesses one disulfide bridge, and the gla residues are located in α helical region. A large volume of in vitro and in vivo studies have documented that OC plays a central role in bone remodeling and skeletal development. More specifically, it activates osteoclasts and recruits their precursors, determining the transition from bone resorption to bone formation [50–53]. Immunohistochemical detection of ON characterizes early stages of bone development, whereas OC typically determines later steps of skeletal growth and osteoid mineralization. Other proteins such as Cbfa1/Runx2 and AP-1 transcription factors have also been found to participate in chondroblastic/osteoblastic differentiation and maturation [54, 55]. Very recent IHC data on rat TMJs have shown that these proteins are selectively expressed in bone and cartilage tissue and that their differential expression highlights different maturation levels during the process of chondro-osteogenesis [56, 57]. Therefore, they support the notion that Runx2 and AP-1 (c-Jun/c-Fos heterodimer) can be used as bone/cartilage markers and indicators of chondro-osteoblastic maturation.
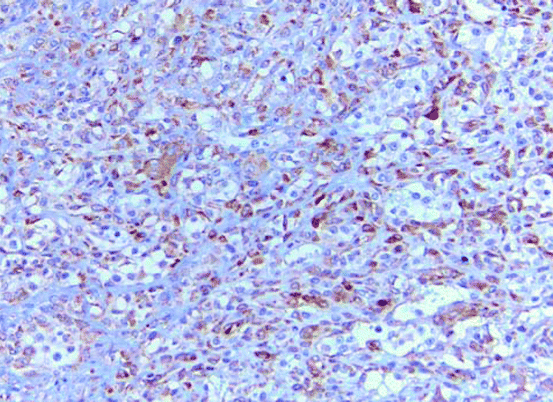
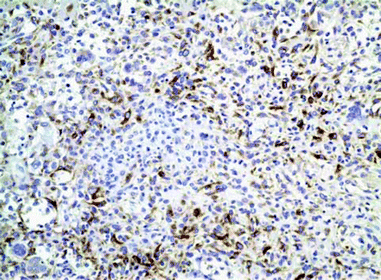
Fig. 8.6
Immunohistochemical method for detection of osteonectin that highlights the osteoid in high-grade osteogenic sarcoma (original magnification ×10)
Fig. 8.7
Same tumor shown in Fig. 8.6, now immunostained for bone-specific marker osteocalcin (original magnification ×10)
8.2.5 In Situ Hybridization
In situ hybridization (ISH) is a valuable molecular method in the field of bone research and diagnosis since it detects the localization of specific nucleic acids at the level of individual cells or complex tissue sections, combining histochemistry with recombinant DNA technology [58–60]. It was first described in 1969 and is based on the specific binding of a labeled nucleotide probe to target DNA or RNA sequences [61, 62]. Probes for ISH (double-stranded DNA, single-stranded antisense RNA, single-stranded DNA probes generated by polymerase chain reaction procedure, synthetic oligodeoxynucleotides, or oligoprobes) are usually 50–300 bases long. Originally, they were labeled with radioisotopes that limited ISH utility for research and diagnostic purposes [60]. Nonetheless, the introduction of non-isotopic labels and development of detection methods based on classical histochemical and immunohistochemical assays reduced the background and improved the signal resolution, expanding the range of ISH applications. In the fields of orthopedic and orthodontic research, ISH can be applicable for both cytogenetic and archival preparations [23, 63–67]. A key advantage of histologic sections is that the examined cells are evaluated in their native architecture and localization. This is often essential, for example, in the study of conditions such as response to stress or metabolic bone diseases, where differential localization of cells with distinct molecular and biochemical properties determines the degree and the quality of bone growth. Furthermore, since locus-specific ISH can be detected by nonfluorescence reagents, it can be easily visualized with bright-field microscopy. The role of fixation is of great importance for ISH since it ensures the integrity of the nucleic acids and the preservation of tissue morphology. Cross-linking fixatives such as formalin and glutaraldehyde seem to provide the best results [68–70]. The degree of DNA/RNA damage caused by decalcification procedures is controversial [64, 65]. However, the combination of EDTA decalcification with formalin fixation seems to be efficient. After fixation and decalcification, unstained sections are transferred onto coated glass slides with advanced adherence properties that prevent tissue floating during the ISH process. Afterward, tissues are treated with pepsin and proteinase K that digest cellular proteins and facilitate probe access to targeted nucleic acids. Optimization of tissue preparation is a major technical challenge. The optimal protein digestion conditions are basically determined empirically for each tissue and probe combination. In order to evaluate whether the signal visualized by ISH is specific for the target DNA or RNA sequence, the use of controls (hybridization of samples from the same tissue with a probe complementary to the assessed probe and identical to the targeted sequence that does not generate any signal) is essential.
The applications of ISH in the areas of bone and dental research are numerous. For instance, it can be performed in order to determine the spatial and temporal distributions of specific mRNA sequences, especially in cases where the gene products are below the threshold of IHC detection. Indeed, by using ISH, the role of genes (namely, Ihh, PTHrP, TRAP, OC, ON, OPN, collagen type II, and Runx1, -2, -3) that are involved in cartilage-bone growth, development, and remodeling has been investigated. Furthermore, ISH has proved to be a valuable tool in the study of pathologic conditions such as metabolic and inflammatory bone and teeth diseases and neoplasias [23, 63, 66, 67, 71, 72]. In situ end-labeling (ISEL) is an ISH-related method that is performed on formalin-fixed, paraffin-embedded tissues for the identification of cells that undergo programmed cell death. ISEL detects the presence of DNA strand breaks that are generated by activated endogenous nucleases during apoptosis [73–75]. More specifically, in the presence of DNA polymerase, the DNA strand breaks are hybridized with non-isotopic reporter molecules, which can be detected with IHC methods. The ISEL assay can be applied as a corollary to the TdT-mediated dUTP-dioxigenin nick-end-labeling (TUNEL) method, which specifically labels the 3′-hydroxyl terminal of DNA strand breaks. Apoptotic cells are recognized by their dark nuclei (TUNEL-positive reaction). During the process of endochondral bone formation, chondrocyts and osteocytes progressively mature and undergo programmed cell death. Osteoclasts are also susceptible to apoptosis in the absence of trophic and growth-stimulating factors, such as M-CSF and RANKL [75, 76]. Obviously, skeletal conditions that induce apoptosis (such as mechanical or biochemical stress and inflammatory or neoplastic diseases) are under intense scrutiny. ISEL and TUNEL are the methods of choice for the investigation of these apoptotic phenomena.
8.3 Polymerase Chain Reaction (PCR) and Reverse Transcriptase PCR (RT-PCR)
The polymerase chain reaction (PCR) assay is a recently developed molecular method for the detection, amplification, and quantization of nucleic acids [77]. Karry Mullis first described it in 1985 [78, 79], winning a Nobel Prize for his achievement. The reagents that are required for PCR include: (a) the dsDNA (template DNA); (b) two PCR primers, which are oligonucleotide sequences of single-stranded DNA that match the sequences at either end of the targeted DNA segment; (c) a thermostable enzyme to synthesize DNA copies; (The most commonly used enzyme is Taq polymerase from the heat-resistant bacterium, Thermus aquaticus.); (d) a pool of four deoxynucleotides (dATP, dCTP, dGTP, dTTP) that will be consumed by polymerase for the synthesis of the new DNA; and (e) buffers containing magnesium that are necessary for the function of Taq polymerase. A single PCR cycle is composed of three sequential steps: denaturation of the dsDNA at 92–96°C, primer annealing or hybridization at 55–72°C, and the synthesis or extension step at 72°C. During the denaturation step, the high temperature facilitates the breaking of hydrogen bonds between complementary bases, resulting in the separation of the dsDNA into two single strands. During the annealing step, the temperature drops, allowing the primers to hybridize to the complementary sequences on the template strands. Usually the primers are 20–30 bases long. The longer the primer, the more specific the binding on the target sequence. During the final step, new DNA strands are produced by Taq polymerase at 72°C. The elongation of the new DNA strands begins by using the oligonucleotide primers as starting points. DNA synthesis progresses from 5′ to 3′ for both new strands. Taq polymerase has the ability to synthesize approximately 1,000 base pairs per minute. The aforementioned three-step procedure is repeated from 30 to 50 times, leading to the synthesis of more than 1 × 109 copies of the original DNA template sequence.
The use of RNA, such as messenger RNA (mRNA), as a template for PCR amplification is accomplished by a modified PCR assay, named reverse transcriptase PCR (RT-PCR) [60, 80]. In a typical RT-PCR, mRNA is extracted from tissue samples or cells and then is copied into DNA (complementary DNA—cDNA) via reverse transcription, which is facilitated by the function of an enzyme called reverse transcriptase. This step is fundamental since Taq polymerase cannot use RNA as a template for the synthesis of PCR products. Several primers (such as random hexamers and oligo-dT) can be used for the reverse transcription step of PCR. The cDNA that is produced serves as the substrate for a classic PCR, as described earlier. In contrast to classic PCR that amplifies genomic DNA (which contains introns and exons), RT-PCR amplifies cDNA (which contains only exons) and therefore only useful genetic information.
The evaluation and analysis of PCR and RT-PCR products are usually made by gel electrophoresis. Electrophoresis is used for the separation of negatively charged nucleic acids, which are mobilized through a liquid or solid matrix by an electric field. Separation is based either on their molecule size or on their three-dimensional conformation. Agarose gel permits the separation of large DNA fragments (1–20 kb), whereas acrylamide gel is optimal for smaller (up to several base pairs) fragments. DNA molecules are visualized by ethidium bromide. The application of PCR in the fields of orthopedics and orthodontics is numerous. More specifically, in the area of bone pathology and oncology, this method can be used for the detection of mutations, polymorphisms, and other genomic alterations in oncogenes and tumor suppressor genes that are involved in tumorigenesis (such as p53, Rb1, p16, HER2/neu, EGF-R, and EXT2) [60]. Regarding skeletal biology, PCR can be used for the detection and quantitative assessment of genes and growth factors that are implicated in several molecular processes, including metabolic bone diseases, tumors, and cellular response to stress factors [81, 82]. Technologic advantages, such as real-time PCR and real-time quantitative TaqMan RT-PCR, are very sensitive, accurate, and highly reproducible methods for the study of gene expression and precise quantification of PCR products [60, 83, 84]. In addition, the development of the in situ PCR assay is an ideal combination of PCR and in situ hybridization that permits the selective amplification and evaluation of specific genetic loci within intact cells [85]. This method can be applied to cells, frozen sections, and sections from archived paraffin-embedded material and has the unique advantage of detecting specific genes within their native environment.
8.4 Microdissection Techniques
Formalin-fixed, paraffin-embedded tissues (FFPET) are some of the most widely available and quality-controlled materials for clinical and basic science studies. However, FFPET are complex, three-dimensional structures, composed of different cell populations with distinct functions. In bone biology, a large volume of studies is focused on the morphologic characteristics and the functional interactions between different cell populations (osteoblasts—osteocytes—chondroblasts). Profoundly, cellular heterogeneity constitutes a major drawback for molecular genetic analyses. Tissue microdissection (TM) represents a reliable method to isolate morphologically well-defined cells and obtain relatively pure cellular populations [81, 83, 84, 86]. DNA or RNA extracted from these populations can be amplified by PCR and then undergo molecular studies for the detection of genetic characteristics and the quantification of genomic alterations. TM can be performed by several different techniques ranging from simple, inexpensive manual TM to more sophisticated (but significantly more expensive) methods such as laser-captured microdissection (LCM).
Manual TM is performed under direct optical visualization of the tissue sample with the use of a stereomicroscope. The target cells are identified on 5-μm thick tissue sections and then dissected with sharp and accurate instruments such as a 30-gauge needle or surgical blade. For better results, tissue sections should undergo deparaffinization prior to microdissection [86]. Tissue fragments are collected in tubes and prepared for nucleic acid extraction and PCR analysis. The primers should be designed to generate small PCR products (<200 bp), since PCR with larger targets may fail. MTM is a fast, cost-effective method that can be applied to any tissue and does not require expensive instrumentation. Nonetheless, neighboring tissues and cells, such as lymphocytes and red blood cells, can very easily contaminate the cell population of interest. In order to obtain uncontaminated cell populations, LCM is the method of choice. LCM was first described by Emmert-Buck in 1999 [87, 88], and several commercially available microdissection systems that use laser technology were developed soon after. The major components of an LCM system are an inverted microscope, an infrared laser, a control unit for the laser, a control mechanism for the microscope stage, a digital camera, and a monitor [89]. LCM can be applied on both frozen and FFPE tissues. Importantly, deparaffinization is required prior to microdissection [90]. The major disadvantage of LCM is the prerequisite of very expensive equipment and well-trained technical stuff. TM is one of the most promising FFPET-based techniques that bridge the gap between morphology and molecular/genetic characteristics.
8.5 Culture of Osteoclasts and Osteoblasts
8.5.1 Osteoclastic Cell Lineage
Bone resorption and bone synthesis are fundamental processes that determine normal bone morphology, skeletal mass, and calcium homeostasis. Any disturbances of this finely tuned interplay result in pathologic conditions such as osteoporosis, osteopetrosis, metabolic bone diseases, fractures, and malignant hypercalcemia. The cells that are specialized to carry out bone resorption are the osteoclasts. Osteoclasts are derived from the pluripotential hematopoietic progenitor CFU-GM (colony-forming unit—granulocyte and macrophage), which also gives rise to monocytes and macrophage-committed precursors [91, 92]. The human mononuclear osteoclast precursor circulates in the monocyte fraction of the peripheral blood. It expresses the monocyte/macrophage integrins CD11b-c and the lipopolysaccharide receptor antigen CD14 [93, 94], as well as the macrophage-associated phenotypes NSE, Mac-1, and Mac-2. In contrast, they are negative for osteoclast-specific markers, namely, tartrate-resistant acid phosphatase (TRAP), vitronectin, and calcitonin receptors [95, 96]. Osteoclast activity is directly and specifically inhibited by calcitonin [97], and therefore receptors that bind calcitonin are considered to be reliable and highly specific markers of mammalian osteoclasts [98]. However, only a small fraction (approximately 2–5 %) of the monocyte/macrophage phenotype cells will eventually differentiate to mature osteoclasts [94]. Under the influence of the transcription factors PU-1 and MiTf, stem cells are committed into the myeloid lineage. In order to progress to the monocyte lineage and express the RANK receptor, M-CSF (macrophage colony-stimulating factor) is required. M-CSF is produced by mesenchymal/stromal cells, including osteoblasts, and is an absolute requirement for the proliferation and differentiation of osteoclast progenitors [99]. M-CSF acts via a tyrosine kinase receptor, named c-fms [100]. Precursors need the presence of RANKL to truly commit to the osteoclast lineage and begin the differentiation program. RANKL is a member of the tumor necrosis factors (TNF) family, which is expressed on the surface of osteoblasts/stromal cells and released by activated T-cells. It binds to RANK receptors on osteoclast precursors and induces their maturation through the nuclear factor-κΒ (NFκΒ) and Jun N-terminal kinase pathways [101, 102]. A member of the tumor necrosis factor receptors superfamily called osteoprotegerin is a decoy receptor for RANKL that inhibits the differentiation and function of osteoclasts [103, 104]. The transition from mononuclear precursor cell to mature osteoclast involves a stepwise loss of macrophage markers and gradual acquisition of phenotypic characteristics specific for osteoclasts. More specifically, since postmitotic osteoclast precursors begin to differentiate into committed osteoclast precursors, they express osteoclast-associated phenotypes, such as TRAP and calcitonin receptors [105]. In contrast, some of the macrophage-related markers, namely, NSE and Mac-1, disappear during osteoclast maturation. Furthermore, they respond to hormones, including 1,25-dihydroxyvitamin D3, parathyroid hormone, and certain cytokines such as IL-1, IL-6, prostaglandins, and colony-stimulating factors. When differentiation of the precursors into pre-osteoclasts is completed, these mononuclear cells begin to fuse, giving genesis to the multinucleated fully mature osteoclasts. Recent evidence suggests that mature osteoclasts undergo apoptosis after a cycle of resorption, a process augmented by estrogens [106].
8.5.2 Culture of Osteoclasts
Since osteoclasts originate from hemopoietic stem cells, bone marrow culture can be used for the study of osteoclast formation. Indeed, Testa and colleagues first demonstrated that multinucleated osteoclasts can be developed in long-term cultures of feline marrow cells [107]. Traditionally, osteoclasts have been generated in cocultures of osteoblasts or stromal cells and hematopoietic cells derived from spleen or bone marrow. For these studies, murine bone marrow cells can be aseptically extracted from long bones of 6–9-day-old mice, following removal of the adhering soft tissues [108]. Afterward, the ends of the bones are removed with scissors, and the bone marrow cells are extracted by slow injection of α-minimum essential medium (α-MEM) into one end of the bone. Bone marrow cells are collected and washed, suspended in α-MEM, and evaluated for viability. Approximately 1 × 107 bone marrow cells can be obtain from a tibia. Coculture methods rely upon the principle that osteoblasts secrete M-CSF and express RANKL after stimulation by 1,25-dihydroxyvitamin D3 and dexamethasone. RANKL binds RANK receptors to monocytic osteoclast precursors, promoting their fusion and thus synthesis of mature multinucleated osteoclasts [101]. Most of the coculture systems occupy UMR-106 rat osteosarcoma cell lines [109].
More recently, following the discovery of RANKL in 1998 [110], it has become possible to generate bone-resorbing osteoclasts without the requirement of osteoblasts in the culture. RANKL ligand and M-CSF can be added directly to osteoclast-precursor cultures, driving the formation of multinucleated, active bone-resorbing cells. This method is easier and more reliable than the coculture method since it employs cells from one single lineage. For osteoclastogenesis experiments that use bone marrow, the extracted cells are washed and cultured in α-MEM with fetal bovine serum (FBS) (10 %), M-CSF (5 ng/mL), and penicillin/streptomycin (1 %) for 48 h. Non-adherent hematopoietic stem cell precursors can be purified with Ficoll-Paque Plus (Amersham Biotech). The interfacial cell layer is isolated for culture in α-MEM with FBS (10 %), M-CSF (30–50 ng/mL), and RANKL (30–100 ng/mL). A TRAP kit can be used for osteoclast staining and counting. Other studies have described the development of osteoclasts from peripheral blood mononuclear cells (PBMNC) [105, 111, 112]. Briefly, 15 mL of blood is mixed with 15 mL of phosphate-buffered saline (PBS) (37°C), purified with 15 mL of Ficoll-Paque, and then centrifuged. Overlying cells are isolated, resuspended in 10 % PBS, diluted with 40 mL PBS, and centrifuged. Isolated PBMNC are placed in a 96-well plate and prewetted by soaking in 100 μL complete α-MEM containing 25 ng/mL M-CSF and 30 ng/mL recombinant RANKL at 37°C. The complete medium should be replaced every 2–3 days. The culture duration for both TRAP staining and pit assays is usually 2–3 weeks.
The multinucleated cells generated by cell cultures can be identified by the presence of certain osteoclast-specific markers: cathepsin K, calcitonin receptor, TRAP, type II carbonic anhydrase, and vitronectin receptor. However, the hallmark of osteoclast identification is the presence of resorption areas on calcified substrates, as defined by osteoclast-resorption lacunae (pit) assays [105, 109, 112].
8.5.3 Osteoblastic Cell Lineage
Osteoblasts arise from pluripotent mesenchymal stem cells, the colony-forming units—fibroblasts (CFU-Fs), which under appropriate stimulation can also give genesis to lipoblasts, chondroblasts, myoblasts, and fibroblasts [113, 114]. The bone morphogenic proteins (BMP) 2–7 and TGF-beta induce the upregulation of transcription factors that mediate the commitment of CFU-Fs toward the osteogenic lineage. The runt homology domain Runx2/Cbfa1 and the zinc finger protein osterix [54, 115, 116] are transcriptional regulators (Fig. 8.8) that facilitate the expression of genes (collagen type I, osteopontin, and alkaline phosphatase) that define the phenotypic features of bone-forming cells. Therefore, these proteins are referred as master regulators of osteoblast morphology. In vivo experiments have documented that knock-out mice lacking Runx2/Cbfa1 and osterix genes do not produce bone [116, 117]. Locally acting proteins, such as platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), and activator protein-1 (AP-1), as well as systemic, blood-circulating molecules, namely, corticosteroids, 1,25 dihydroxyvitamin D3, parathyroid hormone, prostaglandins, and cytokines, are also implicated in specific steps of osteoblastic proliferation and maturation [118].
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


