Fig. 1.1
Schematic depiction of gene signaling patterns in a 7-week-old human fetus. The regional colors represent transient gene expression patterns at different stages of embryogenesis (Courtesy of B. Lozanoff, University of Hawaii
Thus did Vrolik, more than 150 years ago, lay the foundation for understanding the causes of orofacial clefts. The challenge for modern molecular medicine is to translate the clinically observed clefting defects, the phenotype, back through the intricate developmental phenomena that created them, to the coding genotype. The identification of the genetic predisposition to clefts and the environmental factors that determine and alter the varying threshold of normal versus dysmorphic development are among the central challenges for developmental biologists to decipher. Variations of gene expression regulated by epigenetic mechanisms and variable environments may cause differing expressions of genetic traits (polyphenisms), among which are clefting syndromes. The prenatal diagnostic capabilities of chorionic villus sampling, amniocentesis, fetoscopy, magnetic resonance imaging, and ultrasonography have vaulted gestational developmental phenomena into the field of concern to the clinician (Fig. 1.2) (Liou et al. 2011; Stoll and Clementi 2003; Wang et al. 2011). A new three-dimensional sonographic technique (OmniView) allows study of the fetal hard and soft palates and prenatal diagnosis of clefting (Tonni and Lituania 2012). The maldeveloped intrauterine fetus has now become a potential patient (Jones 2002).
Fig. 1.2
Intrauterine ultrasonography of cleft lip fetus (Courtesy of Dr. Eileen Wang, University of Pennsylvania; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
The potential for clefting will ideally be diminished from its initial pathogenetic determination by prevention rather than by post hoc treatment. The basics of biology and molecular medicine will be translated from the laboratory bench to the bedside in the clinical practice of the future.
1.2 Genetics
The mélange of molecular mechanisms involved in the cascading events of embryogenesis are predicated by the expression patterns of specific genes contained in the human genome, constrained by impacting environmental factors. Gene expression patterns are revealing regions of the emerging embryo that have been previously observed histologically and anatomically but not heretofore realized as genetically distinct entities during development. Studies of animal model systems have contributed to our understanding of the molecular determinants that contribute to facial patterning (Swartz et al. 2011). Herein is the marriage of genetics with developmental biology becoming of potential clinical significance.
Current investigations are delineating the complex molecular embryology of development. Specific defects in molecular pathways and networks may provide insights into the etiology of clefting. While embryologists focus on the mechanisms of malformation, deformation, disruption, and dysplasia, clinicians focus on the etiology, diagnosis, treatment, prognosis, and prevention of clefting. The incidence and epidemiology of orofacial clefts provide some clues to the etiology of these malformations (Mossey 2007). The combination of basic and clinical sciences should provide the ideal goal of comprehensive cleft care.
In establishing etiology, it would be useful to have available the gene expression patterns and flow of biochemical pathways underlying morphogenic events. Understanding the local regulation of cellular behaviors and misregulation of any step in these processes can provide insights into clefting consequences. The recognition of the molecular and tissue elements responsible for normal labiopalatogenesis will allow prognostication of clefting defects in their deficiencies. The therapeutic application of growth factors and gene therapy has the potential for biomimetic preventive and healing regimens (Scheller and Krebsbach 2009). Scarless repair of clefts is now potentially feasible (Larson et al. 2010).
Of the estimated 25,000 protein-coding genes in the human genome, some 17,000 genes have been identified in contributing to craniofacial development (International Human Genome Sequencing Consortium 2004). Engineering advancements in genome sequencing technology with the advent of next-generation massively parallel sequencing platforms has made possible the 1,000 Genomes Project which is cataloguing human genotypic variations in all of these genes (Nielsen 2010). The application of whole exome sequencing has proven to be a powerful tool in its ability to identify genes that influence craniofacial patterning (Ng et al. 2010), with whole genome sequencing being the goal of personal medicine for genetic risk assessment and prevention.
The complexity of contributions of the hundreds of genes to facial formation is being elucidated by identifying each gene’s individual expression for each stage of development (Feng et al. 2009). Identification of gene mutations responsible for craniofacial syndromes provides clues to the genetic basis for craniofacial clefting. However, detailed molecular and cellular analyses of the chronology and loci of gene expression patterns, upon which are superimposed epigenetic phenomena, make unraveling the complexity of craniofacial morphogenesis a daunting challenge. The ever-constant new identification of gene expression profiles of embryonic craniofacial and oral structures has led to the development of a consortium titled COGENE (Craniofacial and Oral Gene Expression Network) that can be accessed online at Cogene: http://hg.wustl.edu/COGENE/ (Cai et al. 2005). Therein is contained a list of all hitherto identified genes, growth factors, and signals involved in the expression profiles of structures between the 4th and 8.5 weeks of human development. It is in the mutation or silencing of genes or the misappropriation of growth factors and signals that the source of some developmental defects is revealed. The intricacies of RNA editing, complex regulatory networks, crisscrossing molecular pathways, together with overlapping and redundancies of gene expression patterns make unravelling the skein of individual influences particularly difficult. A FaceBase consortium providing a comprehensive program of craniofacial research has been established (Hochheiser et al. 2011).
Some of the genes implicated in craniofacial development through human and animal model studies are listed in Table 1.1.
Table 1.1
Selected genes implicated in craniofacial development
|
Gene
|
Gene/protein
|
OMIM
|
Associated syndrome (OMIM)/functional role
|
Location
|
|---|---|---|---|---|
|
ALX3
|
Aristaless-like homeobox 3
|
606014
|
Frontonasal dysplasia type 1 (136760)
|
1p13.3
|
|
ARHGAP29/PARG1
|
Rho GTPase-activating protein
|
610496
|
Candidate gene associated with nonsyndromic CL/P
|
1p22.1
|
|
BMP4
|
Bone morphogenetic protein 4
|
112262
|
Orofacial cleft 11 (600625)
|
14q22-q23
|
|
CLPTM1
|
Cleft lip and palate transmembrane protein 1
|
604783
|
t(2;19)(q11.2;q13.3) translocation described in three-generation pedigree with nonsyndrome CL/CP
|
19q13
|
|
CRISPLD2
|
Cysteine-rich secretory protein LCCL domain containing 2
|
612434
|
Candidate gene associated with cleft lip/palate
|
16q24.1
|
|
DLX5
|
Distal-less homeobox 5
|
600028
|
Proximodistal patterning of the pharyngeal arches; oronasal patterning
|
7q21.3
|
|
EFNB1
|
EPH-related receptor tyrosine kinase ligand 2
|
300035
|
Craniofrontonasal dysplasia (304110)
|
Xq13.1
|
|
FAF
|
FAS-associated factor 1
|
604460
|
Pierre Robin sequence, cleft palate only
|
1p32.3
|
|
FGF8
|
Fibroblast growth factor 8
|
600483
|
Kallmann syndrome 6 (612702); cleft lip/palate
|
10q24
|
|
FGF10
|
Fibroblast growth factor receptor 10
|
602115
|
Aplasia of lacrimal and salivary glands (180920); lacrimoauriculodentodigital syndrome (LADD; 149730)
|
5p13-p12
|
|
FGFR1
|
Fibroblast growth factor receptor 1
|
136350
|
Pfeiffer syndrome (101600), Kallmann syndrome type 2 (147950)
|
8p11.23-p11.22
|
|
FGFR2
|
Fibroblast growth factor receptor 2
|
176943
|
Apert syndrome (101200), Pfeiffer syndrome (101600); craniosynostosis and midfacial hypoplasia syndromes; LADD syndrome (149730)
|
10q26.13
|
|
FOXE1 (TTF-2)
|
Forkhead homolog-like 15/thyroid transcription factor 2
|
602617
|
Bamforth-Lazarus syndrome (241850)
|
9q22.33
|
|
GLI2
|
GLI-Kruppel family member 2
|
165230
|
Holoprosencephaly type 9 (610829), cleft lip and palate
|
2q14.2
|
|
GSC
|
Goosecoid
|
138890
|
Homozygous knockout mouse model exhibits aplastic nasal apparatus
|
14q32.1
|
|
GSCL
|
Goosecoid-like
|
601845
|
Located in the DiGeorge syndrome deletion region
|
22q11.21
|
|
HOXA1
|
Homeobox A1
|
142955
|
Athabaskan brainstem dysgenesis (601536); Bosley-Salih-Alorainy syndrome (601536)
|
7p15.3
|
|
IRF6
|
Interferon regulatory factor 6
|
607199
|
Van der Woude syndrome type 1 (119300); popliteal pterygium syndrome (119500); oralfacial cleft 6 (IRF6 enhancer; 608864)
|
1q32.3-q41
|
|
JAG2
|
Jagged2
|
602570
|
Alagille syndrome; association with cleft palate
|
14q32
|
|
LHX6
|
Lim homeobox protein 6
|
608215
|
Expressed in palatogenesis
|
9q33.2
|
|
LHX8
|
Lim homeobox gene 8
|
604425
|
Associated with cleft lip
|
1p31.1
|
|
MEOX2
|
Mesenchyme homeobox 2
|
600535
|
Posterior palate expression in mouse models
|
7p21.2
|
|
MIR140
|
MicroRNA 140
|
611894
|
Regulates PDGFA expression; cleft palate
|
16q22.1
|
|
MSX1
|
Muscle segment homeobox 1
|
142983
|
Oralfacial cleft type 5 (608874); tooth agenesis (106600); Witkop syndrome (189500)
|
4p16.2
|
|
MSX2
|
Muscle segment homeobox 2
|
123101
|
Craniosynostosis type 2; enlarged parietal foramina (168500)
|
5q35.2
|
|
OFC1
|
Unknown
|
119530
|
Oralfacial cleft type 1 (119530)
|
6p24.3
|
|
ORS2
|
Odd-skipped related 2
|
611297
|
Cleft palate in knockout mouse
|
8q22.2
|
|
OTX2
|
Orthodenticle homolog 2
|
600037
|
Anophthalmia, micro-ophthalmia (610125)
|
14q22.3
|
|
PAX7
|
Paired box homeobox gene 7
|
167410
|
Neural crest specification; possible influence on CL/P risk
|
1p36.13
|
|
PDGFC
|
Platelet-derived growth factor C
|
608452
|
Mitogen, associated with CLP
|
4q32.1
|
|
PLCB4
|
Phospholipase C, beta-4
|
600810
|
Auriculocondylar syndrome (602483)
|
20p12
|
|
PTCH1
|
Patched 1
|
601309
|
Holoprosencephaly type 7 (610828), basal cell carcinoma (605462), CLP
|
9q22.32
|
|
PVRL1
|
Poliovirus receptor-like 1/NECTIN-1
|
600644
|
Cleft lip/palate-ectodermal dysplasia syndrome/Zlotogora-Ogur/Margarita Island syndrome (orofacial cleft 7; 225060)
|
11q23.3
|
|
RUNX2
|
Runt-related transcription factor 2
|
600211
|
Cleidocranial dysplasia (119600); implicated in CLP
|
6p21.1
|
|
RYK1
|
Receptor-like tyrosine kinase
|
600524
|
Implicated in oral cleft studies
|
3q22.1
|
|
SATB2
|
Special AT-rich binding protein 2
|
608148
|
Implicated in orofacial clefting
|
2q33.1
|
|
SHH
|
Sonic hedgehog
|
600725
|
Holoprosencephaly type 3 (142945)
|
7q36
|
|
SPRY2
|
Sprouty homolog 2
|
602466
|
Candidate gene for nonsyndromic cleft lip and palate
|
13q31.1
|
|
SUMO1
|
SMT3 suppressor of mif two 3 homolog 1
|
601912
|
Oral facial cleft type 10 (601912)
|
2q33.1
|
|
TCOF1
|
TREACLE
|
606847
|
Treacher Collins-Franceschetti syndrome I (154500)
|
5q32.q33.1
|
|
TBX22
|
T-box 22
|
300307
|
X-linked cleft palate with ankyloglossia (303400)
|
Xq21.1
|
|
TBX10
|
T-BOX 10
|
604648
|
Associated with cleft lip with or without cleft palate
|
11q13.2
|
|
TFAP2A
|
Transcription factor AP-2 alpha
|
107580
|
Branchio-oculo-facial syndrome (113620)
|
6p24.3
|
|
TGFB3
|
Transforming growth factor beta 3
|
190230
|
Cleft palate in mouse model
|
14q24.3
|
|
TGFBR1
|
Transforming growth factor, beta receptor I
|
190181
|
Loeys-Dietz syndrome type 2A (608967), cleft palate
|
9q22.33
|
|
TGFBR2
|
Transforming growth factor, beta receptor II
|
190182
|
Loeys-Dietz syndrome type 2B (610380), cleft palate
|
3p24.1
|
|
TP63
|
Tumor protein p63
|
603273
|
Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 (604292); OFC8 (129400)
|
3q28
|
|
WDR65
|
WD-repeat domain 65;
|
614259
|
Van der Woude syndrome type 2 (606713)
|
1q34.2
|
|
YPEL1
|
Yippee-like 1
|
608082
|
Located in the commonly deleted DiGeorge (188400)/velocardiofacial (192430) region
|
22q.11.2
|
Ascribing specific functions to all these genes is the aim of molecular biology, but it is the realization of the biology encoded within each gene that will provide comprehension of developmental phenomena and their aberrations. Genomic analyses are revealing the molecular architecture behind complex developmental pathways (Brito et al. 2011; Swartz et al. 2011), and the multifactorial basis of the etiology of clefts (Abu-Hussein 2012).
Human genetics can lead to insights of phenotypic diagnosis and provide understanding of the relationships of components of molecular circuitry that will improve the ability of genotypic information to predict the phenotype of complex clefting traits (Dixon et al. 2011; Rahimov et al. 2011; Stuppia et al. 2011; Yuan et al. 2011).
The majority of orofacial clefting cases are nonsyndromic and have no identified cause. Genetic and phenotypic heterogeneity of facial clefting has complicated the identification of the responsible phenomena. Research studies have identified genetic variants associated with cleft lip and palate through linkage analysis, candidate gene approaches, direct sequencing, deletion and duplication analysis by array comparative genomic hybridization (aCGH), and genome-wide association studies (GWAS) (Rahimov et al. 2011; Weatherley-White et al. 2011). A genome-wide association study has identified five genetic loci influencing facial morphology in Europeans: PRDM16, PAX3, TP63, C5o rf 50 and COL17A1 (Liu et al. 2012). This study established links between DNA variants previously associated with non-syndromic cleft lip/palate at 2p21, 8q24, 13q31 and 17q22. The complexity of clefting is illustrated in the numerous different types of genes associated with the phenotype. Such studies have led to the characterization of several genes with variants that convey an increased risk of orofacial clefting including MSX1, a transcription factor expressed in the anterior palate (Jezewski et al. 2003; van den Boogaard et al. 2000), and TGFβ3, a signaling cue involved in cell migration and palatal shelf fusion (Ashique et al. 2002; Iordanskaia and Nawshad 2011; Lidral et al. 1998). TBX22, which functions in conjunction with SUMO1 (Shi et al. 2009) as a transcriptional repressor, participates in posterior palate osteogenesis (Andreou et al. 2007) and whose mutations cause X-linked cleft palate and ankyloglossia (Kantaputra et al. 2011). Another transcription factor is SATB2, which contributes to osteoblastogenesis and influences expression of transcription factors Alx4, Pax9, and Msx1 in the base of the developing palates of mouse models (Britanova et al. 2006; FitzPatrick et al. 2003; Zhang et al. 2011b). The genetic variants contributing to the highest incidence of CL/P (∼2 % of all cases) are found in the interferon regulatory factor 6 (IRF6) gene, a transcription factor that participates in the differentiation of keratinocytes in the developing epidermis (Ingraham et al. 2006; Kondo et al. 2002; Richardson et al. 2006). Mutations or microdeletions in IRF6 are responsible for Van der Woude and popliteal pterygium syndromes, both of which exhibit variable phenotypic expression of labiopalatal clefting and lip pit depressions (Jobling et al. 2011; Kondo et al. 2002). A genome-wide meta-analysis of non-syndromic cleft lip with or without cleft palate has identified six new susceptibility regions viz. 1p36, 2p21, 3p11.1, 8q21.3, 13q31.1 and 15q22 (Ludwig et al. 2012). Variants of SKI have been proposed as a candidate gene for non-syndromic clefts of the lip and palate (Mangold et al. 2012).
Genetically determined elements such as facial and nasal width, bizygomatic distances (Boehringer et al. 2011), palatal height, and jaw growth constitute an additive to the genotype that approaches a cleft palate threshold, whereby each element contributes only a small increase in risk. This threshold may be crossed by external factors that include environmental influences, such as smoking, and potential epidemiological factors including maternal stress and age (Fraser 1976; Jagomagi et al. 2010; Wallace et al. 2011; Wehby et al. 2011; Zhang et al. 2011a; Wu et al. 2012). Maternal smoking during pregnancy implicates TGFB3 and MN1 in the etiology of submucous cleft palate (Reiter et al. 2012).
The functional role of many of these genes has yet to be fully established. Before a link between a gene and its expressed phenotype is recognized, there needs to be extensive characterization of the gene’s products. The topographical areas and timing of gene expression need to be known for specific regions of orofacial development. Interruption of components of the genetic-metabolic machinery responsible for normal embryonic development can lead to malformations. Disharmonic growth between embryonic components occasioned by subtle differences in the number of cell divisions or in the onset or offset times or rates of cellular activities may variably contribute to dysplasias. The biochemical basis of development and growth changes with time during different stages of development.
In a clinically oriented text, consideration of the very early stages of embryogenesis involving molecular biological mechanisms, induction by signaling factors, tissue differentiation, histogenesis, and organ morphogenesis and growth, each of which constitute enormous fields of study, must be greatly condensed. Appreciation of these underlying developmental phenomena is necessary in understanding the series of cascading events leading from the initial zygote formed by the union of parental gametes to the fully fledged infant (Fig. 1.3). Aberrations or variations from the normal morphogenetic patterns, whether of genetic, epistatic, or environmental origin, are responsible for many of the congenital anomalies that constitute clinical syndromes. Currently, there are no specific tests available for genetic susceptibility to orofacial clefts. The revelation of associated congenital anomalies with cleft lip and palate may further identify the interrelationships of diverse embryonic developments with genetic mutations held in common.
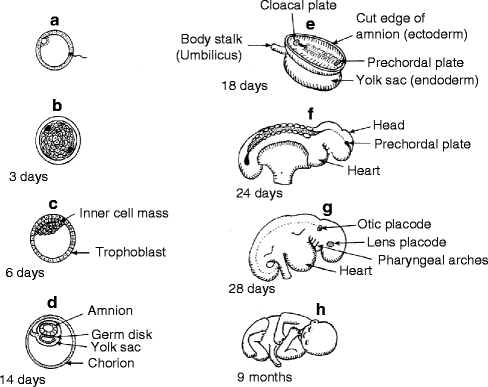
Fig. 1.3
Diagrammatic synopsis of embryogenesis. (a) Spermatozoon penetrating ovum to form zygote. (b) Morula stage of blastula. (c) Blastocyst with inner cell mass. (d) Fetal membranes in chorion. (e) Primary germ layers forming in germ disk. (f) Some stage embryo. (g) Postsomite stage embryo. (h) Full-term fetus (Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
1.3 Early Embryology
The relevance of embryological understanding of facial development is becoming increasingly significant not only for seeking the etiology of orofacial anomalies but also for the application of the molecular mechanisms of normal embryogenesis to the emerging fields of genetic engineering and tissue regeneration. The exploding field of stem cell research for reparative tissue and organ replacement demands an understanding of the morphogenetic mechanisms occurring during facial formation. The recipe for differentiation of stem cells in therapeutic cloning is similar to that of the pathways taken by the multilineage pluripotential cells of the early embryo. The same genes, growth factors, and signaling pathways that operate in the embryo are replicated in directed stem cell differentiation for therapeutic tissue replacement. Fundamental insights into pathophysiology, diseases, and dysmorphology are being revealed by molecular biology (Fig. 1.4).
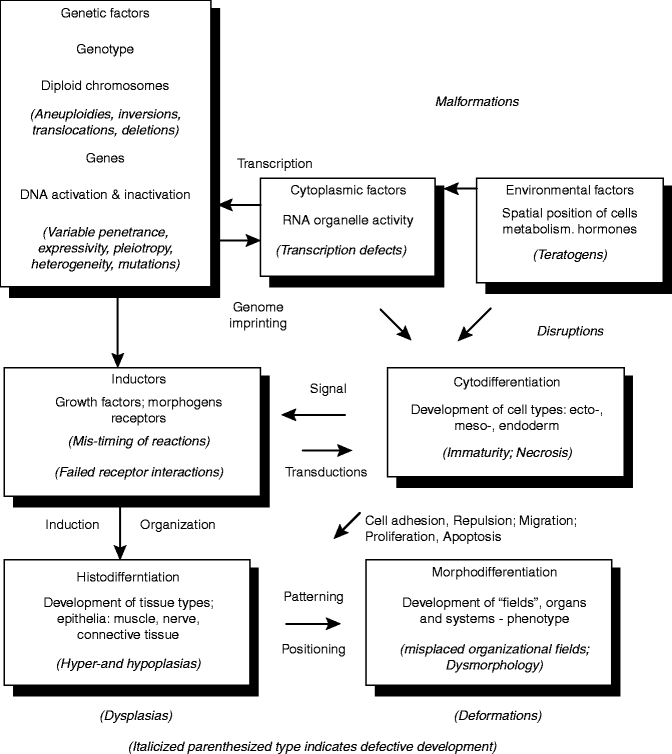
Fig. 1.4
Schema of embryogenesis (Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
The field of craniofacial embryology is currently undergoing a paradigmatic period of readjustment and discovery. The last decade has revealed a host of previously unknown factors in embryogenesis. During development, cells are monitored by genetically determined pathways and adjust their rates of accumulation, apoptosis, and hyperplasia to produce organs of predetermined size. The precise control of growth is of inestimable importance for, if each cell in our faces was to undergo just one more cell division, we would be horribly malformed.
The prior presence of the brain determines the subsequent development of the craniofacies. The rostral parts of the brain—the prosencephalon and mesencephalon—are specified by the orthodenticle homologues OTX1 and OTX2, while the HOX genes specify the rhombencephalon and establish spatial identity of prospective craniofacial compartments (Dixon et al. 2011; Larsen et al. 2010; Vieille-Grosjean et al. 1997). It is the brain underlying the future face that is a key component of cephalogenesis (Marcucio et al. 2011).
1.3.1 Organizing Centers
Human and animal model studies show development of the head depends on the inductive activities of the prosencephalic and rhombencephalic organizing centers, which are regulated by the expression of the sonic hedgehog (SHH) gene as a signaling protein in the neural floor plate cells (De Robertis et al. 1991; Odent et al. 1999). These organizing centers are the sites of origin of signaling factors that diffuse into surrounding areas to create “fate maps” that predetermine the details of differentiation of adjacent cells to form particular facial elements. Thus, the rostral prosencephalic center, derived from prechordal mesoderm, located at the rostral end of the notochord, induces the visual and inner-ear apparatus and upper third of the face (the neurocranium). The caudal rhombencephalic center induces the middle and lower thirds of the face, the viscerofacial skeleton (Fig. 1.5). The gradients of chemical and physical properties emanating from the organizing centers regulate craniofacial patterning by inducing a range of responses from uncommitted populations of neural crest tissue (Hu et al. 2003).
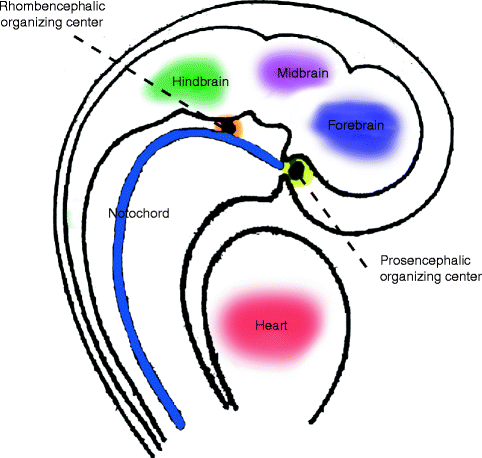
Fig. 1.5
Schematic depiction of prosencephalic and rhombencephalic organizing centers (Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
1.3.2 Neural Crest Tissue
The major contributor to facial formation is the peculiarly derived mesenchymal tissue that arises from the crests of the ectodermal neural folds that create the brain. Specification of the neural crest by the transcription factor PAX7 occurs very early in embryonic development, even before the neural plate appears (Basch et al. 2006; Betters et al. 2010). The transition of the ectoderm into mesenchyme is a key factor in creating ectomesenchyme that provides a lineage of pluripotential cells that gives rise to diverse tissues (Table 1.2). Facial morphogenesis is controlled by multistep reciprocal interactions between the ecto- and endodermal epithelia and neural crest cells.
Table 1.2
Neural crest derivatives
|
Connective tissues
|
|
Ectomesenchyme of facial prominences and pharyngeal arches
|
|
Bones and cartilages of skull and face
|
|
Dermis of face
|
|
Stroma of salivary, thymus, thyroid, parathyroid, and pituitary glands
|
|
Dental papilla, dentin, periodontal ligament, cementum
|
|
Muscle tissues
|
|
Ciliary muscles
|
|
Perimysium, epimysium, endomysium of pharyngeal arch muscles (masticatory, facial, faucial, laryngeal)
|
|
Nervous tissues
|
|
Supporting tissues
|
|
Leptomeninges of prosencephalon and part of mesencephalon
|
|
Glia
|
|
Schwann sheath cells
|
|
Sensory ganglia
|
|
Autonomic ganglia
|
|
Sensory ganglia of trigeminal, facial, glossopharyngeal, and vagal nerves
|
|
Parasympathetic ganglia (ciliary, ethmoid, sphenopalatine, submandibular, enteric system)
|
|
Pigment cells
|
|
Melanocytes in all tissues
|
|
Melanophores of iris
|
The cranial neural crest cells migrate from their initial dorsal location above the rhombomeres of the brain to ventral destinations that are either predetermined by homeobox transcription factor (HOX) genes that constrain their distribution or by responding to local cues from overlying or underlying epithelia (Cordero et al. 2011; Eberhart et al. 2006; Le Douarin et al. 2004; Wilkie and Morriss-Kay 2001). Collagen type I and periostin expression are implicated in the role of cranial neural crest during soft palate development (Oka et al. 2012).
Segmentation of the rhombencephalon into eight rhombomeres delineates the stepwise sequence of cascading streams of migrating ectomesenchyme to create six pharyngeal arches and five facial prominences (Fig. 1.6). Neural crest mesenchyme migrates in the median plane over the prosencephalon to create the frontonasal prominence. Neural crest tissue from the first two rhombomeres migrates ventrally on either side of the rhombencephalon into the first pharyngeal arch that will give rise to both the maxillary and mandibular arches and their derived skeletal elements.
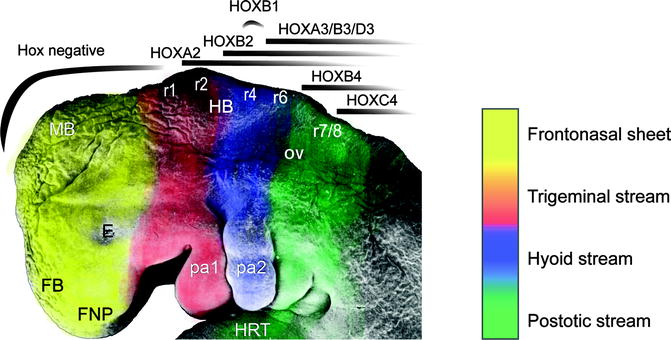
Fig. 1.6
A Stage 15, 33-day-old human embryo upon which are depicted the neural crest streams emanating from the rhombomeres (r1-8), influenced by the homeobox (HOX) gene expression patterns. FNP frontonasal prominence, FB forebrain, E eye, MB midbrain, HRT heart, OV otic vesicle, pa1/2 pharyngeal arches 1/2 (SEM by Prof. Steding, Gottingen. By permission of Springer Verlag; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
Crest tissue from the fourth rhombomere contributes to forming the second pharyngeal arch, while rhombomeres 6 and 7 contribute to the third, fourth, and sixth arches. The neural crest overlying rhombomeres 3 and 5 suffers an apoptotic fate mediated by bone morphogenetic protein-4 (BMP4) signaling before migrating and therefore does not contribute to the arches (Smith and Graham 2001).
Developmental studies have shown platelet-derived growth factor A (PDGFA) is required for normal migration of a subset of neural crest cells toward the oral ectoderm that will participate in palate development (Eberhart et al. 2008). Inadequate neural crest mesenchymal proliferation, migration, or excessive apoptosis would result in deficiencies of tissues, causing clefts, among other hypoplasias (Le Douarin et al. 2004; Noden and Trainor 2005; Wilkie and Morriss-Kay 2001). Treacher Collins syndrome is such an example resulting in apoptosis of the specified cranial neural crest due to disruption of RNA biosynthesis caused by mutations in the TCOF1 gene that encodes for the TREACLE, a nucleolar phosphoprotein (Dixon et al. 2006).
1.3.3 Facial Formation
The orofacial region is identified very early in embryonic development at the 28th day postconception, by the appearance of the prechordal plate in the embryonic trilaminar germ disk. This disk is composed of the three primary germ layers, ecto-, meso-, and endoderm. The prechordal plate is characterized by lack of the intermediate mesoderm. The contiguous ectoderm and endoderm at the site of the prechordal plate combine to form a tenuous and temporary bilaminar oropharyngeal membrane that demarcates the location of the future mouth. The ectoderm will form the mucosa of the future oral cavity, while the endoderm will coat the pharyngeal walls. The oropharyngeal membrane identifies the topographic center of facial development by lining a central depression, the stomodeum, the primitive mouth around which there migrate five facial prominences during the fourth week of embryogenesis (Fig. 1.7). The prescient mouth is bordered rostrally by the developing median frontonasal prominence, laterally by the maxillary prominences, and caudally by the mandibular prominences, the latter two both derived from the first pharyngeal arches (Fig. 1.8) (Sperber et al. 2010).
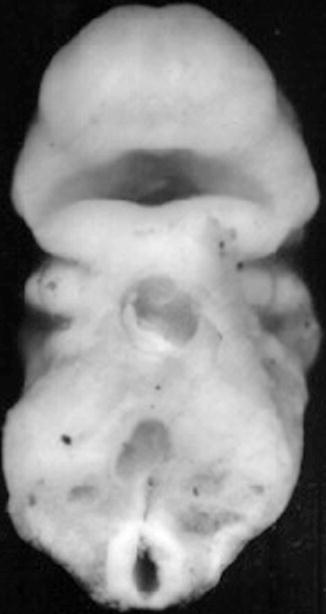
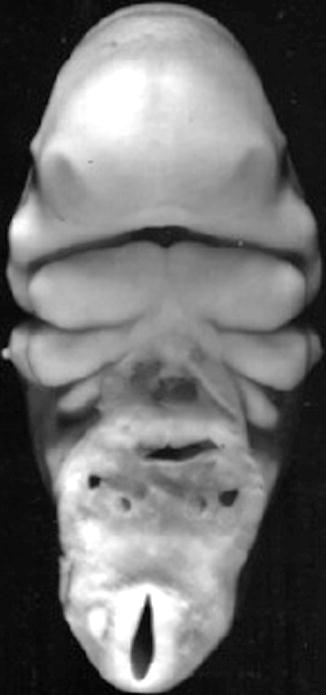
Fig. 1.7
Frontal view of face of a 24-day-old human embryo. ×36 (Courtesy of Prof. Nishimura, Kyoto Collection; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
Fig. 1.8
Frontal view of face of a 32-day-old human embryo. ×22 (Courtesy of Prof. Nishimura. Kyoto Collection; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
The tissues that constitute the frontonasal, maxillary, and mandibular prominences are comprised of cells of different lineages that have migrated, relocated, and been displaced by epithelial-mesenchymal interactions. Neural crest mesenchyme contributes the major tissue type that combines with core mesoderm and is covered by surface epithelia. The neural crest tissues give rise to the facial skeleton, while the mesoderm will form the facial muscles. Four key morphogens control facial development by regulation of cell proliferation, differentiation, survival, and apoptosis (cell death). Extensive studies have shown that these signaling cues include endothelin (ET1) (Clouthier et al. 2010), fibroblast growth factors (FGFs) (Liu et al. 2010; Szabo-Rogers et al. 2008), sonic hedgehog (SHH) (Hu and Marcucio 2009; Welsh and O’Brien 2009), the wingless (WNT) family (Chiquet et al. 2008; Lin et al. 2011; Reid et al. 2011; Mostowska et al. 2012), and the transforming growth factor beta (TGF-β) family (Iwata et al. 2011) which include the bone morphogenetic proteins (BMPs) (Francis-West et al. 2003; Paiva et al. 2010; Spears and Svoboda 2005).
These morphogens direct signaling pathways that interact coordinately and interdependently to regulate the growth, patterning, and shaping of the developing face (Fig. 1.9) (Boehringer et al. 2011; Farlie and Moody 2011; Sperber 2006; Szabo-Rogers et al. 2010). Mutations of genes or misregulation of the signaling pathways results in misappropriated tissue interactions that are the source of facial maldevelopment. The molecular basis for variable expressivity of these genes and factors has not been fully elucidated, but is responsible for the epigenetic spectrum of phenotypic facial malformations. Developmental instability and teratogenic disruption of genetic signaling are other sources of dysmorphic development. Moreover, mechanical pressures must operate within the confines of the epithelial constraints placed upon the expanding mesenchymal components of the facial prominences, influencing their architecture and developing facial features (Radlanski and Renz 2006). A precise mechanistic understanding of the numerous steps involved in signal transductions and migrations is as yet ill-defined.
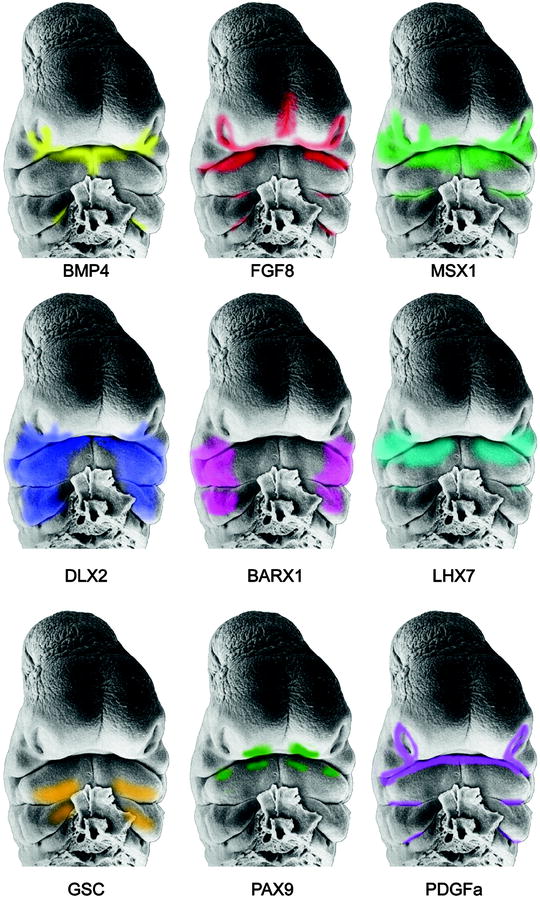
Fig. 1.9
Scanning electron micrographs of the face of a Stage 15, 33-day-old human embryo depicting the gene expression patterns derived from mouse embryos (Faces from Hinrichsen: By kind permission of Springer Verlag; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
The mesodermal core of the first pharyngeal arch condenses into myogenic elements that become innervated by the motor branch of the trigeminal nerve. These muscles migrate to their disparate destinations to perform masticatory and swallowing activities. Similarly, second pharyngeal arch mesodermal myogenic elements, innervated by the facial nerve branches, viz., the occipital, temporal, zygomatic, mandibular, and cervical, migrate through the mesenchymal milieu of the facial prominences to establish all the mimetic muscles of the face (Noden and Francis-West 2006). All these dispersed muscles retain their initially established nerve supply. The lingual musculature is derived from migration and elongation of the hypoglossal cord of somatic mesodermal origin, retaining its original hypoglossal (cranial nerve XII) innervation. Appropriate distribution of all these elements of tissue components will formulate a face of normal physiognomy. Deficiencies of the perioral muscles have been demonstrated in microforms and in full-fledged clefts of the upper lip (Jiang et al. 2006; Landes et al. 2006).
The frontonasal prominence, innervated by the frontal branch of the trigeminal nerve, contributes to the forehead and the nose. On the inferolateral corners of the frontonasal prominence, there develop bilateral nasal placodes that differentiate into the olfactory epithelium that interacts with the underlying olfactory nerves. Defective or absent nasal placodal development not only will result in anosmia but has a devastating effect on nasal and central facial development.
The sinking of the nasal placodes to form nasal pits is the result of the development of the elevated horseshoe-shaped medial and lateral nasal prominences (Fig. 1.10). The posterior aspect of each nasal pit, initially in communication with the stomodeum, becomes separated from the oral cavity by the transient oronasal membrane. This membrane normally disintegrates by the end of the 5th week postconception to open the posterior choanae connecting the nostrils to the posterior oral cavity. Failure of membrane disintegration leads to choanal atresia, a potentially fatal asphyxiating neonatal congenital anomaly.
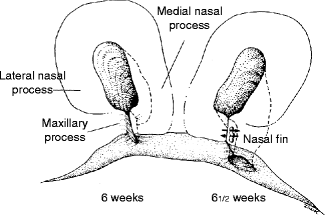
Fig. 1.10
Schematic depiction of breakdown of nasal fin and formation of nostrils. Arrows indicate disintegration of the nasal fin between the medial nasal and maxillary prominences (Courtesy of J. Avery and Oxford University Press; Reproduced by kind permission of McGraw-Hill from Losee and Kirschner (2009))
Elevation of the lateral nasal prominences creates the alae of the nose. The expression patterns of 36 genes are manifested in the medial nasal prominences and those of some 45 genes in the lateral nasal prominences. The location of these genes can be identified on the gene resource locator (Ref: http://grl.gi.k.u-tokyo.ac.jp
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


