http://evolve.elsevier.com/Haveles/pharmacology
A drug is a biologically active substance that can modify cellular function. A general understanding of drug action is important and will allow the dental hygienist to make informed decisions regarding possible drug interactions or adverse reactions for the patient. This chapter discusses the action of drugs in the body and methods of drug administration. Chapter 3 considers the problems or adverse reactions these drugs can cause. By understanding how drugs work, what effects they can have, and what problems they can cause, the dental hygienist can better communicate with the patient and other health care providers about medications the patient may be taking or may need to have prescribed for dental treatment.
Characterization of drug action
Dose-response curve, potency, and efficacy are terms used to measure drug response or action.
Log Dose Effect Curve
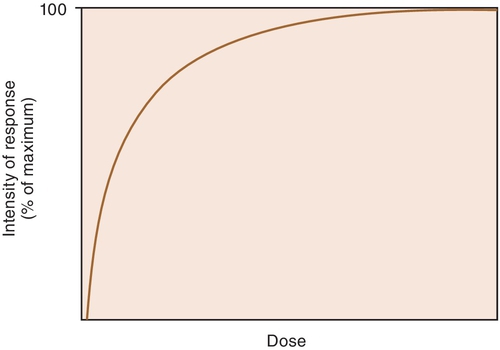
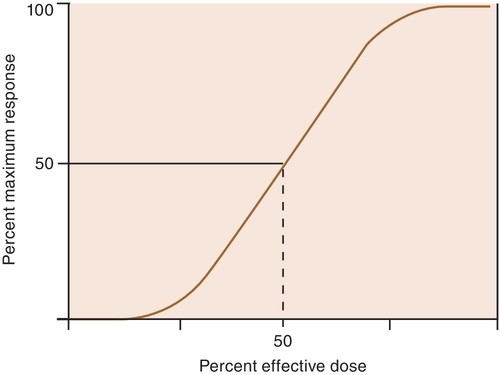
When a drug exerts an effect on biologic systems, it is possible to measure the response to the dose of the drug given. If the dose of the drug is plotted against the intensity of the effect, a curve will result (Figure 2-1). If this curve is replotted using the log of the dose (log dose) versus the response, another curve is produced from which the potency and efficacy of a drug’s action may be determined (Figure 2-2).
Potency
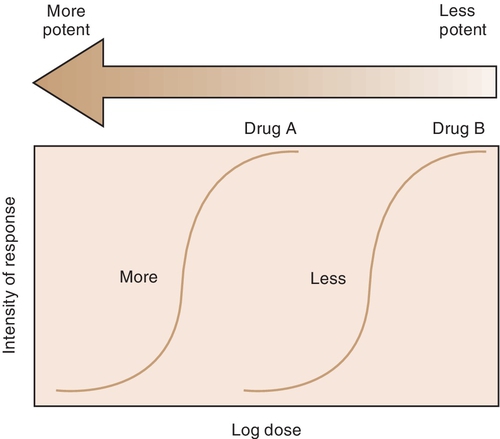
The potency of a drug is a function of the amount of drug required to produce an effect. The potency of a drug is shown by the location of that drug’s curve along the log-dose axis (x-axis). The curves in Figure 2-3 illustrate two drugs with different potencies. The potency of drug A is greater because the dose required to produce its effect is smaller. The potency of B is less than that of A because B requires a larger dose to produce its effect.
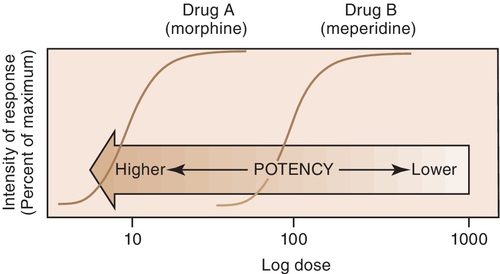
For example, both meperidine and morphine have the ability to treat severe pain, but approximately 100 mg of meperidine would be required to produce the same action as 10 mg of morphine. In Figure 2-4, the curve for drug B (meperidine) is to the right of the curve for drug A (morphine) because the dose of meperidine needed to produce pain relief is larger (10 times larger) than that of morphine. The potency of the drug should not be an issue as long as appropriate doses of medication are used. Less potent drugs require higher doses to produce therapeutic effects, whereas more potent drugs can reach toxic levels at lower doses. Review the proper dose of each drug before it is prescribed to the patient.
Efficacy
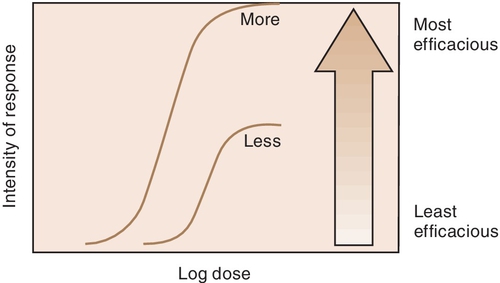
Efficacy is the maximum intensity of effect or response that can be produced by a drug. Administering more drug will not increase the efficacy of the drug but can often raises the probability of an adverse reaction. The efficacy of a drug increases as the height of the curve increases (Figure 2-5). The efficacy of the drugs whose curves are illustrated in Figure 2-5 are shown by the height of the curve when it plateaus (levels out horizontally). It is shaded from least (light) to most (dark) potent. The efficacy of any drug is a major descriptive characteristic indicating its action. For example, the efficacy of drug B (meperidine) and that of drug A (morphine) are about the same because both drugs relieve severe pain.
Therapeutic Index
The therapeutic index (TI) is a ratio of the median lethal dose (LD50) to the median effective dose (ED50) and is expressed as follows:
TI=LD50/ED50
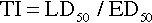
Because death is the end point when one is measuring the lethal dose, LD50 is the dose that causes death in 50% of test animals. The ED50 is the dose required to produce the desired clinical effect in 50% of test animals. The greater the TI, the safer the drug. Drugs with a lower TI (closer to zero) require careful monitoring to avoid toxic reactions. An example of a drug with a low TI is digoxin, a drug used to treat heart failure.
Mechanism of action of drugs
After drugs have been distributed to their site of action, they elicit a pharmacologic effect. The pharmacologic effect occurs because of a modulation in the function of an organism. Drugs do not impart a new function to the organism; they merely produce either the same action as an endogenous agent or block the action of an endogenous agent. This signaling mechanism has two functions: amplification of the signal and flexible regulation. The presence of very fine controls to modulate the body’s function allows the regulation of certain reactions, slowing or speeding them.
Receptors
Once a drug passes through the biologic membrane, it is carried to many different areas of the body, or site of action, to exert its therapeutic effect or adverse effect. For the drug to exert its effects, it must bind with the receptor site on the cell membrane. Drug receptors appear to consist of many large molecules that exist either on the cell membrane or within the cell itself (Figure 2-6). More than one receptor type or identical receptors can be found at the site of action. Usually, a specific drug binds with a specific receptor in a lock-and-key fashion. Many drug-receptor interactions consist of weak chemical bonds, and the energy formed during this interaction is very low. As a result, the bonds can be formed and broken easily. Once a bond is broken, another drug molecule immediately binds to the receptor.
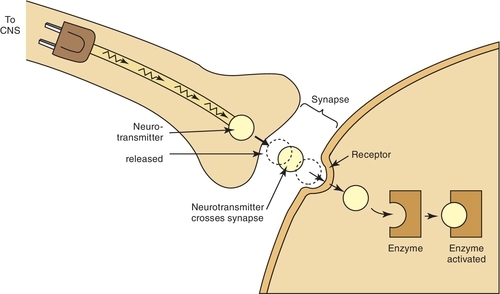
Different drugs often compete for the same receptor sites. The drug with the stronger affinity for the receptor will bind to more receptors than the drug with the weaker affinity (Figure 2-7). More of the drug with the weaker affinity will be required to produce a pharmacologic response. Drugs with stronger affinities for receptor sites are more potent than drugs with weaker affinities for the same sites.
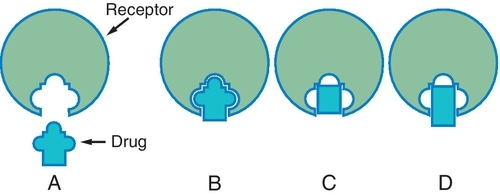
Agonists and Antagonists
When a drug combines with a receptor, it alters the function of the organism. It may produce enhancement or inhibition of the function. Drugs that combine with the receptor may be classified as either agonists or antagonists (Figure 2-8).
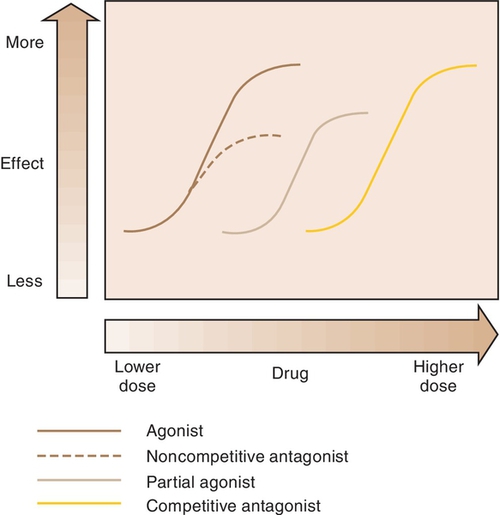
Agonist
An agonist is a drug that (1) has affinity for a receptor, (2) combines with the receptor, and (3) produces an effect. Naturally occurring neurotransmitters are agonists.
Antagonist
An antagonist counteracts the action of the agonist. The following are three different types of antagonists:
Pharmacokinetics
Pharmacokinetics is the study of how a drug enters the body, circulates within the body, is changed by the body, and leaves the body. Factors that influence the movement of a drug are divided into four major steps: absorption, distribution, metabolism, and excretion (ADME).
Passage Across Body Membranes
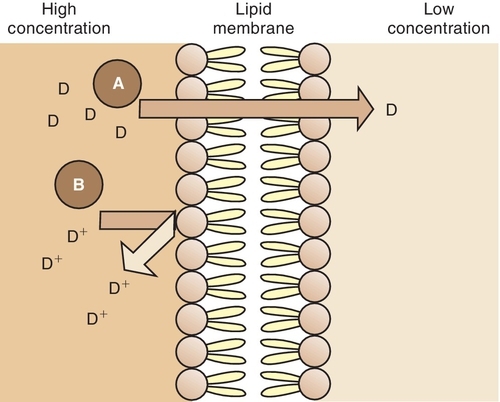
The amount of drug passing through a cell membrane and the rate at which a drug moves are important in describing the time course of action and the variation in individual response to a drug. Before a drug is absorbed, transported, distributed to body tissues, metabolized, and subsequently eliminated from the body, it must pass through various membranes, such as cellular membranes, blood capillary membranes, and intracellular membranes. Although these membranes have variable functions, they share certain physicochemical characteristics that influence the passage of drugs across them.
These membranes are composed of lipids (fats), proteins, and carbohydrates. The membrane lipids make the membrane relatively impermeable to ions and polar molecules. Membrane proteins make up the structural components of the membrane and help move the molecules across the membrane during the transport process. Membrane carbohydrates are combined with either proteins or lipids. The lipid molecules orient themselves so that they form a fluid bimolecular leaflet structure with the hydrophobic (lipophilic) ends of the molecules shielded from the surrounding aqueous environment and the hydrophilic ends in contact with the water. The various proteins are embedded in and layered onto this fluid lipid bilayer, forming a mosaic. Studies of the ability of substances to penetrate this membrane have indicated the presence of a system of pores or holes through which lower-molecular-weight and smaller chemicals can pass.
The physicochemical properties of drugs that influence the passage of drugs across biologic membranes are lipid solubility, degree of ionization, and molecular size and shape. The mechanisms of drug transfer across biologic membranes are passive transfer and specialized transport.
Passive Transfer
Lipid-soluble substances move across the lipoprotein membrane by a passive transfer process called simple diffusion. This type of transfer is directly proportional to the concentration gradient (difference) of the drug across the membrane and the degree of lipid solubility. For example, a highly lipid-soluble compound attains a higher concentration at the membrane site and readily diffuses across the membrane into an area of lower concentration (Figure 2-9). A water-soluble agent has difficulty passing through a lipoprotein membrane.
Specialized Transport
Certain substances are transported across cell membranes by processes that are more complex than simple diffusion and filtration. These processes are as follows:
Absorption
Absorption is the process by which drug molecules are transferred from the site of administration to the circulating blood. This process requires the drug to pass through biologic membranes.
The following factors influence the rate of absorption of a drug:
Effect of Ionization
Drugs that are weak electrolytes dissociate (separate) in solution and equilibrate into an un-ionized form and an ionized form. The un-ionized, or uncharged, portion acts like a nonpolar, lipid-soluble compound that readily crosses body membranes (see Figure 2-9). The ionized portion will pass across these membranes with greater difficulty because it is less lipid soluble.
The pH of the tissues at the site of administration and the dissociation characteristics (pKa) of the drug will determine the amount of drug present in the ionized and un-ionized state. The proportion in each state will determine the ease with which the drug will penetrate the tissues.
Weak Acids
When the pH at the site of absorption increases, the hydrogen ion concentration simply falls. This results in an increase in the ionized form (A−), which cannot easily penetrate tissues.
Conversely, if the pH of the site falls, the hydrogen ion concentration will rise. This results in an increase in the un-ionized form (HA), which can more easily penetrate tissues.
Weak Bases
If the pH of the site rises, the hydrogen ion concentration falls. The drop results in an increase in the un-ionized form (B), which can more easily penetrate tissues. Conversely, if the pH of the site falls, the hydrogen ion concentration rises, resulting in an increase in the ionized form (BH+), which cannot easily penetrate tissues. In summary, weak acids are better absorbed when the pH is less than the pKa, whereas weak bases are better absorbed when the pH is greater than the pKa.
This dissociation also explains the fact that in the presence of infection, the acidity of the tissue increases (and the pH decreases) and the effect of local anesthetics decreases. In the presence of infection, the [H+] increases because of accumulating waste products in the infected area. The increase in [H+] (decrease in pH) leads to an increase in ionization and a decrease in penetration of the membrane. This decreased penetration reduces the clinical effect of the local anesthetic.
Oral Absorption
The dose form of a drug is an important factor influencing absorption of drugs administered via the oral route. Unless the drug is administered as a solution, the absorption of the drug in the gastrointestinal tract involves a release from a dose form such as a tablet or capsule. This release requires the following steps before absorption can take place:
Absorption from Injection Site
Absorption of a drug from the site of injection depends on the solubility of the drug and the blood flow at that site. For example, drugs with low water solubility, such as some penicillin salts, are absorbed very slowly after intramuscular injection. Absorption at injection sites is also affected by the dose form. Drugs in suspension are absorbed much more slowly than those in solution. Certain insulin preparations are formulated in suspension form to decrease their absorption rate and prolong their action. Drugs that are least soluble will have the longest duration of action.
Distribution
Basic Principles
All drugs occur in two forms in the blood: bound to plasma proteins and the free drug. The free drug is the form that exerts the pharmacologic effect. The bound drug is a reservoir (place to store) for the drug. The proportion of drug in each form depends on the properties of that specific drug (percentage protein bound). Within each compartment (e.g., blood, brain), the drug is split between the bound drug and the free drug. Only the free drug can pass across cell membranes.
For a drug to exert its activity, it must be made available at its site of action in the body. The mechanism by which this is accomplished is distribution, which is the passage of drugs into various body fluid compartments such as plasma, interstitial fluids, and intracellular fluids. The manner in which a drug is distributed in the body will determine how rapidly it produces the desired response; the duration of that response; and in some cases, whether a response will be elicited at all.
Drug distribution occurs when a drug moves to various sites in the body, including its site of action in specific tissues. However, drugs are also distributed to areas where no action is desired (nonspecific tissues). Some drugs, because of their characteristics, are poorly distributed to certain regions of the body. Other drugs are distributed to their sites of action and then redistributed to other tissue sites. The distribution of a drug is determined by several factors, such as the size of the organ, the blood flow to the organ, the solubility of the drug, the plasma protein-binding capacity, and the presence of certain barriers (blood-brain barrier, placenta).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


