Fig. 6.1
Photograph of the teeth of a 13-year-old boy with hypoplastic AI showing pitted enamel surfaces
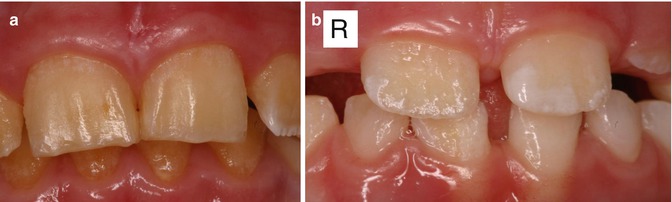
Fig. 6.2
(a) Photographs of the maxillary incisors of a boy from a family with X-linked amelogenesis imperfecta showing minimal enamel present. (b) Incisors of the sister of the boy in (a) showing pits, irregular ridges of normal enamel, and areas of absent enamel. These features are more noticeable on the right-side incisor
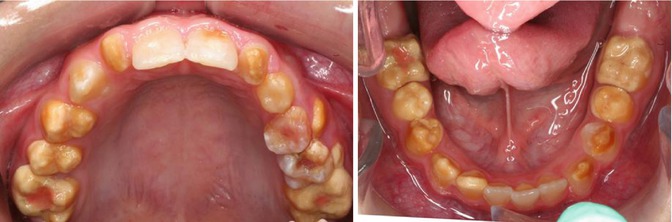
Fig. 6.3
Photographs showing the maxillary and mandibular teeth of an 11-year-old boy with hypocalcified AI
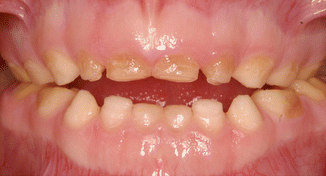
Fig. 6.4
Photographs of a 5-year-old boy’s primary dentition that is affected by hypomaturation type of AI showing anterior open bite
Genotypes and Phenotypes of Amelogenesis Imperfecta
Recent advances in molecular genetics and biochemistry have made it possible to subtype the AI phenotypes based on the type of genetic mutation. The AI mutations and proteins associated with some of the hypoplastic, hypocalcified, and hypomaturation phenotypes and their modes of inheritance are shown in Tables 6.1 and 6.2, while the OMIM designations and genes involved in some AI conditions are shown in Table 6.3. To date, only approximately half of all AI phenotypes are thought to be caused by mutations in known genes that affect enamel formation, namely, AMEL, ENAM, FAM83H, KLK4, MMP20, WDR72, and C4ORF26, while the genetic changes involved in the other half of AI phenotypes are currently unknown [6, 71, 91].
Table 6.1
Phenotypes and genotypes of hypoplastic types of amelogenesis imperfecta
|
Phenotype
|
Inheritance
|
AI mutation
|
Protein
|
Authors
|
|---|---|---|---|---|
|
Hypoplastic – smooth
|
AD
|
ENAM
|
p.A158Q178del
|
Rajpar et al. (2001) [57]
|
|
Hypoplastic – thin
|
AD
|
ENAM
|
p.N197fsX277
|
Kida et al. (2002) [28]
|
|
Hypoplastic
|
AD
|
ENAM
|
p.M71_Q157del
|
Kim et al. (2005) [31]
|
|
Hypoplastic – localized
|
AD
|
ENAM
|
p.K53X
|
Mardh et al. (2002) [46]
|
|
Hypoplastic – localized
|
AD
|
ENAM
|
p.422fsX277
|
Hart et al. (2003) [21]
|
|
Hypoplastic – localized
|
AD
|
ENAM
|
p.S246X
|
|
|
Hypoplastic – smooth, thin
|
AR
|
ENAM
|
p.V340_M341insSQYQYCV
|
|
|
Hypoplastic – smooth, thin
|
AR
|
ENAM
|
p.P422fsX448
|
Hart et al. (2003) [21]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.15_a8delinsT
|
Lagerstrom-Fermer et al. (1991) [38]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.W4X
|
Sekiguchi et al. (2001) [62]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.MIT
|
Kim et al. (2004) [30]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.W45
|
Kim et al. (2004) [30]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.E191X
|
Lench and Winter (1995) [41]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.P158fsX187
|
Lench and Winter (1995) [41]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.L181fsX187
|
Kindelan et al. (2000) [33]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.Y147fsX187
|
Green et al. (2002) [19]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.H129fsX187
|
Sekiguchi et al. (2001) [62]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
p.P52R
|
Kida et al. (2007) [29]
|
|
Hypoplastic – smooth
|
X-linked
|
AMEL
|
pT511
|
Lench and Winter (1995) [41]
|
Table 6.2
Phenotypes and genotypes of hypocalcified and hypomaturation types of amelogenesis imperfecta
|
Phenotype
|
Inheritance
|
AI mutation
|
Protein
|
Authors
|
|---|---|---|---|---|
|
Hypomaturation/hypoplastic
|
X-linked
|
AMEL
|
p.18del
|
Lagerstrom et al. (1991) [37]
|
|
Hypomaturation/hypoplastic
|
X-linked
|
AMEL
|
p.H77L
|
Hart et al. (2003) [21]
|
|
Hypomaturation
|
X-linked
|
AMEL
|
p.P701
|
Collier et al. (1997) [10]
|
|
Hypomaturation
|
AR
|
MMP20
|
p.1319fs338X
|
Kim et al. (2005) [31]
|
|
Hypomaturation
|
AR
|
MMP20
|
p.H226Q
|
|
|
Hypomaturation
|
AR
|
MMP20
|
p.W34X
|
Papagerakis et al. (2008) [53]
|
|
Hypomaturation
|
AR
|
WDR72
|
p.Ser783X
|
El-Sayed et al. (2009) [13]
|
|
Hypomaturation
|
AR
|
WDR72
|
p.S489fs498
|
Wright et al. (2011) [91]
|
|
Hypomaturation
|
AR
|
WDR72
|
p.Lys333X
|
Kuechler et al. (2012) [35]
|
|
Hypomaturation
|
AR
|
KLK4
|
p.W153X
|
Hart et al. (2004) [22]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.S287X
|
Wright et al. (2009) [88]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.Q470X
|
Wright et al. (2009) [88]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.Q456X
|
Hart et al. (2009) [23]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.L308fsX323
|
Wright et al. (2009) [88]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.W460X
|
Lee et al. (2008) [39]
|
|
Hypocalcified
|
AD
|
FAM83H
|
p.Q677X
|
Lee et al. (2008) [39]
|
|
Hypocalcified – localized
|
AD
|
FAM83H
|
p.L625fsX703
|
Wright et al. (2009) [88]
|
|
Hypocalcified – localized
|
AD
|
FAM83H
|
p.E694X
|
Wright et al. (2009) [88]
|
Table 6.3
Hereditary conditions with enamel defects – OMIM designations and genes
|
Amelogenesis imperfecta
|
Gene/locus
|
Enamel phenotype
|
Mode of inheritance
|
|---|---|---|---|
|
# 301200. Amelogenesis imperfecta, type IE; AI1E
|
AMELX
|
Hypoplasia/hypomaturation depending on mutation and protein effect
|
X-linked
|
|
% 301201. Amelogenesis imperfecta, hypoplastic/hypomaturation, X-linked 2
|
Xq22-q28
|
Hypoplastic and/or hypomaturation
|
X-linked
|
|
#104500. Amelogenesis imperfecta, type IB; AI1B
|
ENAM
|
Localized hypoplastic/generalized hypoplastic
|
Autosomal dominant
|
|
#204650. Amelogenesis imperfecta, type IC; AI1C
|
ENAM
|
Generalized hypoplastic
|
Autosomal dominant
|
|
#204700. Amelogenesis imperfecta, hypomaturation type, IIA1; AI2A1
|
KLK4
|
Normal enamel thickness – hypomineralized orange-brown color
|
Autosomal recessive
|
|
#612529. Amelogenesis imperfecta, hypomaturation type, IIA2; AI2A2
|
MMP20
|
Normal enamel thickness – hypomineralized orange-brown color
|
Autosomal recessive
|
|
#130900. Amelogenesis imperfecta, type III; AI3
|
FAM83H
|
Localized or generalized hypomineralized enamel
|
Autosomal recessive
|
|
#613211. Amelogenesis imperfecta, hypomaturation type, IIA3; AI2A3
|
WDR72
|
Hypomaturation – creamier/opaque enamel upon eruption. Discoloration and loss of tissue post-eruption
|
Autosomal recessive
|
|
#104510. Amelogenesis imperfecta, type IV; AI4
|
DLX3
|
TDO – thin pitted hypoplastic
|
Autosomal dominant
|
|
# 614253. Amelogenesis imperfecta and gingival fibromatosis syndrome; AIGFS
|
FAM20A
|
Generalized hypoplastic and failure of tooth eruption, gingival hypertrophy
|
Autosomal recessive
|
|
%104530. Amelogenesis imperfecta, hypoplastic type
|
???
|
Hypoplastic – failure to erupt and calcification of pulp. 6 different forms
|
?
|
|
#614832. Amelogenesis imperfecta, hypomaturation, IIA4; AI2A4
|
C4ORF26
|
Hypomaturation AI
|
Autosomal recessive
|
Mutations in the gene coding for the enamel protein ENAM have been reported for two distinct types of AI that are mostly transmitted in an autosomal dominant (AD) manner [28, 31] where the inheritance rate of AI from an affected parent is 1:2 regardless of gender. In addition, a few families with ENAM mutations have reported autosomal recessive (AR) transmission [21, 51] where the rate of inheriting AI from two carrier parents is 1:4 regardless of gender (Table 6.1). ENAM mutations are associated with hypoplastic phenotypes with the enamel defects presenting as generalized thin or pitted enamel [89]. Some ENAM mutations, e.g., in p.K53X [46], are associated with localized defects, most likely from haploinsufficiency (reduced enamel production due to decreased amounts of enamelin that is being produced from the one normal allele). Other types of ENAM mutations, e.g., p.N197fsX277 [28], may result in thin or absent enamel, probably as a result of production of abnormal proteins that are nonfunctional for enamel formation.
The AMELX gene encodes for the enamel protein amelogenin which has key roles, which are as yet not fully understood, in the enamel extracellular matrix that undergoes mineralization [72]. As approximately 90 % of human amelogenin is expressed from the AMELX gene, and only 10 % from the AMELY gene, inheritance of most AMEL mutations, is X-linked and is characterized by males typically having a more severe phenotype compared with females [83]. The rate of inheriting the X-linked gene is 1:2 for both male and female children of an affected mother and normal father. For children of an affected father and normal mother, the inheritance rate is 1:2 for female children and none of the male children will inherit the gene. Females with the AMELX gene mutations typically show the Lyonization effect where partial X-chromosome inactivation in the ameloblasts results in alternating vertical bands of normal and abnormal enamel [82, 83]. In contrast, affected males usually show a generalized and more severe enamel phenotype compared with females as they are only producing the abnormal protein from the abnormal AMELX gene (Fig. 6.2).
Mutations of the AMELX gene result in phenotypes that have been reported as hypoplastic or hypomaturation phenotypes (Tables 6.1 and 6.2). Generalized thin hypoplastic AI phenotypes can result from AMELX mutations in the C-terminus-coding regions as well as from abnormalities in the formation of signal peptides [38, 39, 61]. In contrast, enamel hypomaturation phenotypes are associated with mutations in the N-terminus-coding region of AMELX [20, 37], while mutations in exons 6 and 5 cause a combined hypomaturation-hypoplastic phenotype [2, 29, 40].
Mutations in the FAM83H gene result in autosomal dominant hypocalcified AI that is thought to be the most common form of AI in the USA [88]. Although the role of FAM83H in other tissues is unclear, the fact that all FAM83H mutations reported to date are associated with enamel changes points to the significance of FAM83H in enamel formation. Individuals with FAM83H mutations show weak, yellow brown discoloration of the enamel with severely reduced mineral and increased protein content which contrasts with that of hypomaturation AI in being not proline rich [88]. The generalized types of hypocalcified AI affecting the entire crowns result from FAM83H mutations that are usually associated with nonfunctional proteins (e.g., p.Q677X). In contrast, the localized types which are caused by mutations associated with less dysfunctional proteins (e.g., p.E694X) result in enamel changes that are seen mainly in the cervical parts of the crowns [88].
Mutations in the genes coding for the proteinases kallikrein-4 (KLK4) and metalloproteinase MMP20 cause hypomaturation phenotypes and are inherited as autosomal recessive traits [42, 86]. The enamel is hypomineralized with a high protein content although the thickness is normal. Kallikrein-4 (KLK4) codes for a proteinase that removes remaining proteins during the maturation phase to allow for optimal crystal growth and mineralization [86]. MMP20 metalloproteinase is required for cleaving amelogenin and ameloblastin during the secretory stage of enamel formation. Multiple allelic mutations in MMP20 have been identified [18, 32, 52, 53], and the resulting phenotypes are different from the autosomal recessive pigmented hypomaturation AI that is caused by the C4ORF26 gene mutations. Another group of mutations associated with autosomal recessive hypocalcified or hypomaturation types of AI is found in mutations of the gene WDR72 that codes for an intracellular protein thought to have mediatory functions between proteins [17, 85].
Other Abnormalities Associated with Amelogenesis Imperfecta
Other oral conditions are encountered in patients with AI more frequently compared to the general population. The most well known of these is skeletal open bites which occur commonly with the hypomaturation and hypocalcified phenotypes (Fig. 6.4) [9, 25, 54]. The etiology of skeletal open bites in AI is unclear, but has been speculated to be associated with effects of the genetic changes in other tissues. It is possible that anterior open bites may also result from abnormal jaw posturing or changes in bite force due to severe dental sensitivity of the AI teeth [9]. Other abnormalities that have been associated with AI include taurodontism, eruption delay/failure, hypercementosis, pulp calcifications [44], impaction of teeth, and follicular cysts [9, 65, 80].
Other Hereditary Conditions with Enamel Defects
Molecular defects can alter enamel development by a variety of mechanisms and there are thousands of genes expressed by ameloblasts supporting their activities toward enamel development. Genetic alterations can exert a direct effect through gene expression by the enamel-forming cells (e.g., ameloblasts secrete an abnormal matrix such as in AI) or by secondary effects where the gene may not be dysfunctional or expressed by the ameloblast (e.g., underlying mesenchymal cells affected or systemic metabolic alteration). Genetic mutations can have a direct effect on the oral epithelium, thereby altering the differentiation or function of the ameloblasts or adjacent supporting cells (e.g., stratum intermedium). For example, junctional epidermolysis bullosa (OMIM 226700, 226650) is associated with enamel hypoplasia secondary to abnormal laminin 5 formation that is critical for cell attachment [1, 87]. Most hereditary conditions affecting enamel formation result in a hypoplastic enamel phenotype. For example, junctional epidermolysis bullosa (JEB) caused by alteration of laminin 5 has a thin and/or pitted enamel phenotype. Because laminin 5 also is a critical component of the epidermal-dermal junction, skin fragility is a hallmark feature of JEB. Depending on the severity of skin fragility, the oral health management of individuals with JEB can be very challenging.
Other conditions such as tricho-dento-osseous syndrome (TDO – OMIM # 190320) that is caused by mutations in the transcription factor DLX3 also have a thin and/or pitted enamel phenotype. Affected individuals are almost always born with kinky-curly hair (half of them lose this characteristic by childhood) and dense cranial and skeletal bone that becomes apparent on radiographs during childhood [90]. There is marked variability in affected individuals despite almost all of the cases having the same genetic mutation in the DLX3 gene. Taurodontism and large pulp chambers with thin dentin are a common feature of this condition and help delineate it from AI. The propensity for developing pulp necrosis and having abscess formation appears to stem from the combination of thin enamel, large pulps, and thin dentin that allow microexposure of the dental pulp and bacterial invasion. Placing resin copings or crowns to help prevent pulp exposure can be beneficial in the primary and developing permanent dentition in more severely affected individuals.
There are numerous forms of ectodermal dysplasias that can have significant enamel defects such as Goltz syndrome (also called focal dermal hypoplasia – OMIM # 305600) and ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome (OMIM # 604292) to name just a couple. While most syndromes are associated with a hypoplastic enamel phenotype, the severity of the enamel defect varies markedly, and there can be many associated features such as hypodontia and facial clefting as examples. Therefore, the management of each of these conditions will be different depending on the nature of the enamel defects (e.g., hypoplastic and/or hypomineralized) and the presence of associated oral and systemic conditions.
Management of Patients with Hereditary Defects of Enamel Development
Clinical problems commonly experienced by many patients affected with developmental defects of enamel, regardless of whether they are associated with AI or a syndrome, are compromised esthetics, dental sensitivity, tooth wear, and increased risk for caries and calculus formation [47, 64]. Management of enamel defects will be predicated on understanding and accounting for the associated oral and systemic manifestations and potential need for special medical management (e.g., treatment of severe EB conditions). The severity of the enamel defects and associated problems varies depending on the specific condition. For example, with the AI conditions the hypocalcified and hypomaturation variants have more severe signs and symptoms compared with the hypoplastic types. The aims of dental treatment are to reduce dental sensitivity, improve estethics, restore masticatory function, and prevent deterioration of the dentition from caries, fracture, tooth wear, and erosion. Early diagnosis and preventive care are essential for effective preservation of the dentition. Clinicians should also be alert to the fact that psychological distress and low self-esteem are prevalent among AI children, particularly those with the more severe types, presumably from the reduced quality of life associated with poor esthetics and tooth sensitivity [8, 24]. Children who have a family history of AI or medical syndromes that are commonly associated with defects of enamel development such as epidermolysis bullosa should be examined as soon as the primary and permanent teeth emerge [66]. Referral to specialist pediatric dentists, pediatricians, and geneticists for definitive diagnosis, genetic testing, and counseling may be required.
In clinical practice, management of patients with developmental enamel defects may be considered in a few age-related phases: 1–6 years old (primary dentition or initial phase), 6–12 years old (early permanent dentition or transitional phase), and teenagers and adults (full permanent dentition). Treatment planning is likely to be complex and, for children and adolescents, usually involves interdisciplinary specialists including general dental practitioners, specialist pediatric dentists, and orthodontists. A prosthodontist may need to be consulted when the children reach adulthood to manage the complex prosthodontic treatment that is usually required. The preservation of tooth structure should be a central strategy for all patients with enamel defects, and dental management should aim at preventing caries, maintaining good gingival health, and restoring the teeth with minimally invasive treatment options. Maintaining the dentition and alveolar bone is critics so that future implants or prostheses will have optimal bony support.
Preventive Care
For all children with defects of enamel development, the restorative, prosthetic, and orthodontic treatments should be supported by an aggressive program of preventive care. Oral hygiene instruction, diet counseling, topical fluoride therapy, and frequent periodic examinations should be instituted immediately after diagnosis. Preventive care is crucial as children’s enamel defects such as AI are at high risk for caries and periodontal disease due to the presence of rough defective enamel surfaces that often extend subgingivally and render oral hygiene difficult. In addition, many children, particularly those with hypocalcified and hypomaturation AI phenotypes, have a higher tendency for calculus formation, probably due to changes on the enamel surfaces, saliva, or plaque microbial flora [84]. Gingival overgrowth may occur as an exaggerated response to retained bacterial plaque and mechanical irritation of abnormal enamel that may further compromise esthetics [45]. As AI children often have difficulties with oral hygiene due to tooth sensitivity, toothbrushing and flossing techniques should be taught to the parents and child and reinforced frequently. Short recalls for professional scaling and cleaning are recommended for children who are unable to perform oral hygiene thoroughly [47
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


