Fig. 7.1
Photographs and an OPG of a patient with type II dentinogenesis imperfecta showing amber primary and permanent teeth with tooth surface loss (enamel fracture and attrition of underlying dentin). The OPT shows short, narrow roots with pulpal obliteration of the primary teeth and the first permanent molars
This is the isolated nonsyndromic type of DGI and is caused by mutations in the DSPP gene. The DSPP gene codes for the most abundant of the non-collagenous proteins in dentin.
Clinical presentation
The dental features of this type are similar to those of DGI-I, but penetrance is virtually complete (normal teeth are never found). There might be some variation in the severity of the condition between different members of the same family and individual teeth in the same patient. In addition, the crowns are typically bulbous with marked cervical constriction. The teeth tend to be clinically smaller than unaffected teeth.
Radiographical presentation
Similar to DGI-I.
Dentinogenesis Imperfecta (Shields Type III)
This is also known as the Brandywine isolate type as it was first described in a triracial population from Maryland and Washington DC.
Clinical presentation
This type has variable expressivity with clinical features sometimes resembling those of DGI-I and type II.
Radiographical presentation
The primary teeth appear hollow radiographically due to the lack of dentin formation and very large pulp chambers. As a result of the large pulp chambers, enamel fracturing and rapid dentin wear, these teeth develop multiple pulp exposures and are prone to abscess formation.
Dentin Dysplasia (Shields Type I – OMIM)
Clinical presentation
The dental crowns in DD-I appear generally normal in shape, form and colour. However, the dentin has a pathognomic cascading waterfall histological appearance in thin sections, and the root form is abnormal to varying degrees. The teeth often are displaced or malaligned and have increased mobility. These teeth have an increased risk for developing periapical abscesses in the absence of dental caries or other aetiological factors.
Radiographical presentation
The roots can be sharp with conical, apical constrictions or have extremely short blunt roots. Pre-eruptive pulpal obliteration occurs in both the primary and permanent teeth. Pulpal obliteration is usually complete in the primary teeth; however, it is often only partial in the permanent dentition (crescent-shaped pulpal remnant parallel to the cementoenamel junction). Numerous periapical radiolucencies are often seen in non-carious teeth (Fig. 7.2).
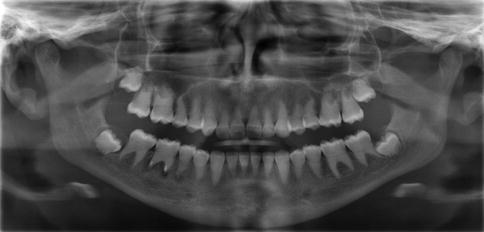
Fig. 7.2
This individual with DD type I has complete obliteration of all the pulp chambers that is seen to be occurring even in the unerupted third molars. The roots are short, and the mandibular right first permanent molar has developed periapical pathology
Dentin Dysplasia (Shields Type II – OMIM 125420)
Clinical presentation
The features seen in the deciduous dentition resemble those observed in DGI-II; however, the permanent dentition is either unaffected or shows mild radiographic abnormalities and can have slight colour changes.
Radiographical presentation
Thistle-tube deformity of the pulp chamber of the permanent teeth and frequent pulp stones.
Molecular Aetiology
The organic component of dentin is composed predominantly of type I collagen (85 %) and non-collagenous protein mainly dentin phosphoprotein (50 %). Numerous genes are involved in regulating the production of the complex dentinal extracellular matrix which eventually mineralises in a highly controlled way.
Type I collagen is the product of COL1A1 and COL1A2 genes, while dentin phosphoprotein and dentin sialoprotein (DSPP) are products of the DSPP gene. There are known to be hundreds of different mutations in COL1A1 and COL1A2 genes that are associated with osteogenesis imperfecta; however, only some of the collagen mutations result in dentinogenesis imperfecta. Mutations in either type 1 collagen genes or the DSPP gene can alter the essential interactions between these proteins resulting in abnormal mineralisation and a DI dental phenotype.
Syndromic Dentin Defects
Osteogenesis Imperfecta
This condition is usually a result of mutations in one of the two collagen type 1 genes (COL1A1 and COL1A2). This condition is transmitted as an autosomal dominant or autosomal recessive trait, and there are multiple types of OI. The most common classification [8] lists four subtypes, but there are now over ten listed on OMIM. The severity varies markedly between different individuals and families. These OI conditions are characterised by bone fragility, blue sclera, deafness mainly at third decade of life and lax ligaments (hypermobility of joints). In severe cases, there can be marked bony deformities, including scoliosis, compression fractions of the vertebrae and ribs, short stature, frontal bossing and other manifestations. Dentinogenesis imperfecta can be a manifestation of osteogenesis imperfecta.
Many individuals with severe forms of OI will be treated with intravenous bisphosphonate treatments to try and improve bone density and decrease the risk of fractures. While IV bisphosphonate treatment has been associated with bone necrosis following dental extractions, there have not been cases of this problem reported in OI patients.
Ehlers-Danlos Syndrome (EDS)
Ehlers-Danlos syndrome is the most common of the heritable connective tissue disorders. It consists of nine major variants with inheritance patterns of either autosomal dominant or recessive, depending on the type. The clinical manifestations in the different EDS conditions result from defects in the synthesis, secretion or polymerisation of collagen. The main features of EDS include tissue fragility, skin extensibility and joint hypermobility.
Defective dentinogenesis affecting the mandibular incisors was reported in some EDS type I where aplasia or hypoplasia of root development, bulbous enlargement of the roots and pulp stones were reported [9]. Others reported dental features such as dysplastic dentin and obliterated pulp chambers [10].
EDS type VIII (OMIM # 130080) is associated with severe early-onset periodontitis that is managed with aggressive local debridement and antibiotic therapy.
Goldblatt Syndrome (OMIM # 184260)
Goldblatt syndrome, also known as spondylometaphyseal dysplasia, is characterised by joint laxity, mesomelic limb shortening and DGI. The deciduous teeth display typical features of DGI, but the permanent teeth appear normal [11].
Schimke Immuno-Osseous Dysplasia (OMIM # 242900)
Schimke immuno-osseous dysplasia is an autosomal recessive disorder caused by mutations in the SMARCAL1 gene. It is characterised by a combination of spondyloepiphyseal dysplasia, progressive renal disease and lymphopenia with defective cellular immunity [11]. Patients with this condition were also reported to have dental features of DGI, such as a grey-yellowish discoloration of the dentin, bulbous crowns with a marked cervical constriction and small or obliterated pulp chambers [11].
Hypophosphataemic Rickets, X-Linked Dominant Vitamin D-Resistant Rickets (OMIM # 307800)
X-linked dominant hypophosphataemic rickets is one of the types of rickets where patients have bone deformities, short stature and hypophosphataemia. Hypophosphataemia is the result of mutations in the phosphate-regulating gene on the X chromosome that results in renal wasting of phosphorus at the proximal tubule level. These patients, therefore, are characterised by low renal phosphate reabsorption, normal serum calcium level with hypocalciuria, normal or low serum level of vitamin D (1,25(OH)2D3, or calcitriol), normal serum level of PTH and increased activity of serum alkaline phosphatases [7, 12].
Dentally, the dentine is the most markedly affected tissue in these patients. They present clinically with dental abscesses in the absence of caries, radiographically with larger pulp chambers and elongated pulp horns and histopathologically with interglobular dentine. Dental attrition can lead to exposure of the large pulp horns causing loss of vitality and abscesses [7, 13].
Management of Defects in Dentin Formation
The management of individuals with defects of dentin can be challenging from both diagnostic and treatment perspectives. Obtaining the diagnosis is based on careful evaluation of the family history, clinical manifestations and radiographic appearance. A genetics consultation should be considered for dentin defects that appear to be hereditary. Management often requires immediate-, short- and long-term planning. The long-term management of these patients usually requires an interdisciplinary approach starting in the primary/early mixed dentition.
Patients with dentinal defects of primary teeth often present at a young age with parents wanting immediate treatment to improve the child’s appearance or because the teeth are beginning to develop abscesses. However, it is important to make a treatment plan which will encompass a stepwise approach that includes a vision to help achieve the short-, medium- and long-term treatment goals of a functional and aesthetic dentition. Children with dentinal defects often require several interventions throughout their primary, mixed and possibly permanent dentition stages. Should general anaesthesia (GA) be required to provide extensive restorative/surgical treatments, it is crucial to plan and provide the necessary management in a timely manner so that the least number of GAs are required throughout the child’s life.
The goals of oral health management, especially at the primary dentition stage, include the following:
1.
Obtaining a diagnosis of the condition if it is unknown
2.
Prevention and maintenance of good oral hygiene
3.
Desensitisation and pain management of affected teeth (if required)
4.
Restorative management (aesthetic management and stabilisation of affected teeth)
5.
Behaviour management to help children manage the dental care they need
General Management
Need for Early Detection
Early detection of tooth structural defects such as DGI, DD and amelogenesis imperfecta is of great importance for multiple reasons. These conditions can be associated with other more serious conditions such as osteogenesis imperfecta and vitamin D deficiency which would allow early detection of these conditions. Additionally, some of these conditions are associated with tooth surface, possible pulp exposure or periapical infections, therefore; early detection would allow close monitoring of these cases and early intervention and prevention where needed.
Care should be taken not to confuse these cases with other conditions with similar clinical presentation such as [2]:
1.
Amelogenesis imperfecta where enamel defects causes enamel loss and exposure of underlying dentin (Fig. 7.3)
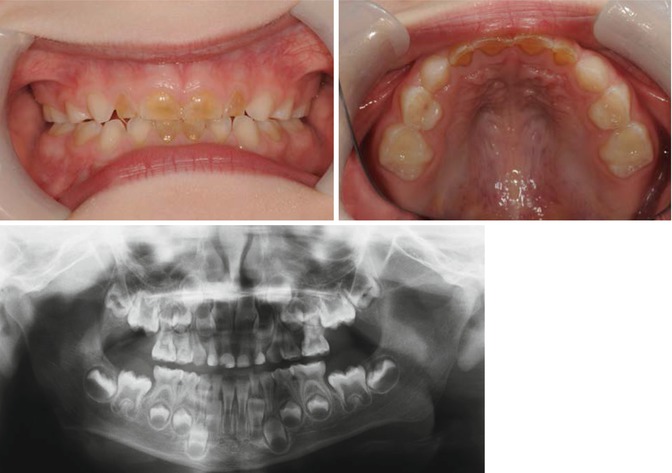
Fig. 7.3
Photograph of a patient with hypomineralised amelogenesis imperfecta with amber upper and lower anterior primary teeth. OPT showed no difference in radiodensity of enamel and dentin on all anterior teeth and all Ds in addition to reduced radiopacity of the enamel on posterior teeth suggestive of amelogenesis imperfecta
2.
Intrinsic discolouration resembling amber/translucent colour:
(a)
Red-brown discolouration due to haemolytic anaemia such as congenital erythropoietic porphyria and rhesus incompatibility
(b)
Yellow or grey to brown discolouration such as tetracycline stain
(c)
Green discolouration: Hyperbilirubinaemia such as in cases of congenital biliary atresia (Fig. 7.4), acute liver failure and biliary hypoplasia
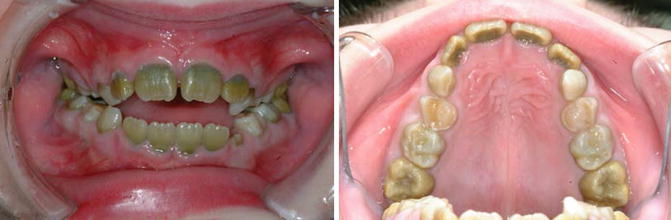
Fig. 7.4
Photographs of a patient with biliary atresia showing green discolouration of primary and permanent teeth due to hyperbilirubinaemia
3.
Mobility leading to early tooth loss resembling teeth lost in DGI-III and DD-I due to periapical abscesses and short roots such as:
(a)
Hypophosphatasia
(b)
Immunological deficiencies, e.g. severe congenital neutropenia, cyclic neutropaenia, Chediak-Higashi syndrome, neutropaenias, histiocytosis X, Papillon-Lefevre syndrome and leucocyte adhesion deficiency syndrome
Assessment of Medical History
A medical history should aim to establish if the dental condition is a ‘syndromic’ form of DGI as this is a variable feature of a number of heritable conditions as discussed previously. For instance, patients with DGI should be checked for history of bone fractures with minimal trauma, joint hyperextensibility, short stature, hearing loss and scleral hue in order to help exclude osteogenesis imperfecta. This is especially important in what appear to be sporadic cases of DGI that have no family history.
This is important as the overall dental management of these patients should take into account other associated medical conditions such as OI, hypophosphataemic rickets, EDS, etc. For instance, patients with EDS can have different complications depending on the type of EDS such as fragile skin tissues, structural heart defects, poor wound healing, hypermobility of the joints and fragile blood vessels. It is therefore important to liaise carefully with the patient’s physician in order to determine the exact type and the associated risks prior to arranging any dental treatments.
Patients with OI and hypophosphataemic rickets are susceptible to bone fractures, and therefore, physical restraint is contraindicated in these patients. In addition, manual handling of these patients under GA should be performed with extreme care.
Furthermore, over the last decade bisphosphonates are being increasingly used in children and adolescents especially for the management of OI. Although bisphosphonates are well tolerated, concerns exist regarding bisphosphonate-related osteonecrosis of the jaw or BRONJ. BRONJ has been defined as ‘a condition of exposed bone in the mandible or maxilla that persists for more than 8 weeks in a patient who has taken or currently is taking a bisphosphonate and who has no history of radiation therapy to the jaw’ [14
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


