The infant brain develops at a rapid rate during the first few months of life. The cerebral volume doubles during the first 6 months and doubles again by the first year of life. By age 2 years, the average child has attained about 80% of the adult brain size, and this significant growth over the first few months of life requires a dynamic yet protective skull. Under normal conditions, the brain volume triples within the first year of life and by the second birthday, the cranial capacity is 4 times that at birth.
Cranial sutures represent a form of articulation between plates of membranous bone by a thin layer of fibrous tissue. Six major sutural areas constitute the cranial vault ( Fig. 1 ), which allow head deformation during vaginal delivery. During postnatal development, cranial vault sutures allow rapid skull expansion and accommodate the significant brain expansion. Minimal pressure (approximately 5 mm Hg) from the growing brain is required to stimulate new bone deposition at the margins.
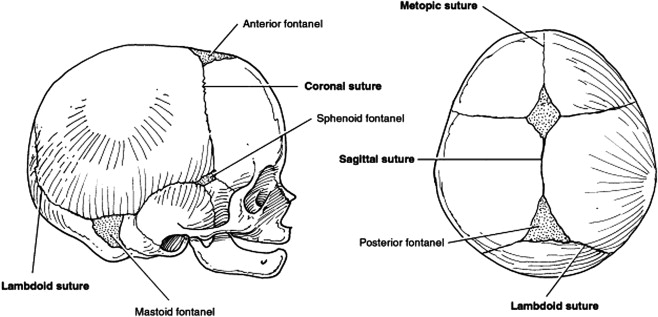
Craniosynostosis is defined as a premature fusion of the cranial vault suture or sutures, which often occurs during fetal development. Because it is an intrauterine event, a more accurate description may be congenital absence of the cranial vault sutures. Craniosynostosis results in fusion of the adjacent bones with arrested sutural growth in the affected region. Virchow’s classic theory states that premature fusion of a cranial vault suture results in limited development of the skull perpendicular to the fused suture and a compensatory “overgrowth” through the sutures that remain open. The result is a characteristic dysmorphology related to the affected suture, with potential neurologic consequences if underlying brain compression occurs. Most forms of craniosynostosis represent random, nonsyndromic malformations limited to the cranial vault and orbital regions.
In this article, current diagnostic and surgical treatments for nonsyndromic craniosynostosis are presented, and perioperative considerations with specific surgical maneuvers used to treat different types of craniosynostosis are outlined with clinical examples. Management of these patients requires a combined neurosurgical and craniofacial team approach for thorough evaluations of the cranial abnormality. Well-planned surgical procedures are required to release the involved suture and reshape the dysmorphic skeletal components, and these timely interventions restore the skeletal architecture, allowing unrestricted brain growth.
Functional consequences of craniosynostosis
During the first 2 years of life, the cerebral volume increases at a rapid rate, while growth of the visceral structures (ie, brain and eyes) at the same time causes growth of the skeletal structures (cranial vault and orbits). A complex of flexible cranial bones created by patent cranial vault sutures allow the growing brain to push them outward. At these open regions, cranial growth occurs in a perpendicular direction to each suture and by ectocranial deposition of bone (and simultaneous endocranial resorption).
Intracranial Pressure
Growth along an affected cranial suture is diminished when it is fused, and the compression of a rapidly expanding brain within the limited intracranial volume may result in increased intracranial pressure (ICP, usually >15 mm Hg). This sequence of events is traditionally used to explain the theoretical relationship between craniosynostosis and brain insult, but it does not adequately explain why only certain patients develop elevated ICP. Previous investigations have determined that approximately 14% of children with untreated single-suture craniosynostosis demonstrate increased ICP. When 2 or more sutures are fused, the likelihood of elevated ICP increases to 42%, the functional consequence of craniosynostosis of most concern, because it may adversely affect brain function.
Children with untreated craniosynostosis who develop increased ICP may exhibit several neurologic symptoms, including headaches, vomiting, sleep disturbances, feeding difficulties, behavioral changes, and diminished cognitive functioning. However, such symptoms related to ICP are difficult to detect before one year of age, often presenting with a slow, gradual onset and, left untreated, may be irreversible. In some instances, early recognition of developmental anomalies is difficult because of the lack of proper testing measures.
Hydrocephalus
Hydrocephalus is encountered in approximately 10% of children with multiple-suture craniosynostosis and often seen in the craniofacial dysostosis syndromes. In contrast, hydrocephalus is not usually observed in patients with nonsyndromic single-suture craniosynostosis, but it may occur independently and not necessarily as a consequence of this condition. For cases in which the infant presents with hydrocephalus and craniosynostosis, careful consideration must be given to the exact timing and sequence of the cranial vault reshaping and ventriculoperitoneal (VP) shunt placement. Often, the placement of the VP shunt should be performed as a separate surgery, after the cranial vault reconstruction when possible (using a temporary ventricular drain for the surgical procedure if necessary). Constant cerebrospinal fluid diversion via a VP shunt may decrease brain expansion, reduce the physical support to the bone segments, and, in some instances, alter the skull shape.
Ophthalmologic Effects
The optic nerve is at risk in patients with craniosynostosis and associated elevated ICP. Papilledema, optic nerve atrophy, and eventual loss of vision (possibly complete) may occur with prolonged, untreated ICP elevations. Visual function and eye motility are often impaired when orbital deformity occurs. For example, orbital dystopia secondary to unilateral coronal synostosis can result in disturbances of extraocular muscle movement (ie, strabismus), upper eyelid ptosis, and poor binocular vision. Decreased orbital volume (as seen in cases of syndromic or nonsyndromic bilateral coronal craniosynostosis) causes proptosis, corneal exposure, and increased risk of direct ocular trauma.
Diagnostic approach to abnormal head shape
When an infant presents with a cranial vault asymmetry, the examiner must be alert to the possibility of prematurely fused sutures. However, abnormal head shape may be produced by various causes, which must be distinguished from craniosynostosis. Most commonly, external forces applied to a flexible cranium with open sutures causes a deformation of the skull. The baby’s early descent into the true pelvis (often a narrow birth canal) may result in deformational plagiocephaly involving the anterior and posterior cranial vault. Postnatal and external forces from repetitive sleep positioning may cause these self-limiting skull shape abnormalities. In 1992, the American Academy of Pediatrics recommended that infants sleep on their backs to reduce the risk of sudden infant death syndrome. An increased number of referrals for nonsynostotic posterior plagiocephaly has resulted because of this “Back to Sleep” campaign and the increased awareness of cranial vault deformities within the pediatric community.
Proper identification of cranial vault deformities resulting from nonsynostotic causes (ie, positional plagiocephaly) and those associated with a true absence of a cranial vault suture (ie, craniosynostosis) is critical for diagnosis and appropriate treatment. The importance of a correct diagnosis is underscored by the dramatically different management approaches for each infant. Craniosynostosis requires surgical treatment, whereas benign positional skull molding requires conservative nonsurgical management using custom-made cranial orthotic devices and is not associated with neurologic or developmental sequelae.
A careful history and clinical examination are the primary basis for diagnosis of craniosynostosis, because each form produces a unique head shape. Because the condition is an intrauterine event, parents often report that the deformity was noted immediately after birth and without subsequent improvement. By contrast, cases of positional plagiocephaly have well-rounded head shape at birth, with asymmetry developing 3 to 6 months later (suggesting postnatal deformation of a flexible cranial vault). A consistent combination of arrested growth in one region and compensatory increase in others may explain unilateral sutural involvement causing a bilateral deformity or posterior suture fusion producing an anteroposterior (A-P) dimension deformity. An examiner familiar with these aberrant growth patterns and the characteristic dysmorphologies specific to each type of synostosis should consistently establish an accurate clinical diagnosis.
Craniosynostosis is primarily diagnosed by careful clinical examination but must be confirmed radiographically. Following clinical evaluation by a craniofacial team, a complete skull series of plain radiographs is obtained to evaluate the cranial vault sutures (usually not needed for the simple suture synostosis). Standard views of the cranial vault are often adequate to establish the absence of a cranial vault suture and confirm the diagnosis. For cases in which all the sutures cannot be adequately visualized using plain films, additional imaging using computed tomography (CT) is indicated. In addition to confirming the diagnosis, high-quality craniofacial CT scans provide more detailed 3-dimensional morphologic information, which is useful during the surgical planning phase. One-millimeter axial and coronal cuts with overlap and 3-dimensional reconstructions of the cranial vault, cranial base, orbits, and maxillofacial skeleton are recommended. Precise patient positioning is important in obtaining a diagnostic scan, and this may require sedation or general anesthesia.
Although technological advancements have dramatically increased the accuracy of standard radiographic studies and CT imaging, incorrect and outdated terminology is still occasionally used in describing cranial vault sutures. At some medical centers, radiologists and surgeons continue to use terms such as fibrous synostosis, suture dysfunction, and impending synostosis when describing cranial vault sutures that otherwise appear open. It is known that craniosynostosis is most often an intrauterine event that is usually recognized at birth. Therefore, a patent suture on a diagnostic-quality radiographic study negates the diagnosis of craniosynostosis. Although the postnatal fusion of cranial vault sutures has been described in the scientific literature, it is considered an extremely rare event that is specifically related to certain clinical and syndromic conditions. Some terms not consistent with contemporary knowledge about the condition may be confusing to parents and health care providers, perhaps resulting in inappropriate management.
Diagnostic approach to abnormal head shape
When an infant presents with a cranial vault asymmetry, the examiner must be alert to the possibility of prematurely fused sutures. However, abnormal head shape may be produced by various causes, which must be distinguished from craniosynostosis. Most commonly, external forces applied to a flexible cranium with open sutures causes a deformation of the skull. The baby’s early descent into the true pelvis (often a narrow birth canal) may result in deformational plagiocephaly involving the anterior and posterior cranial vault. Postnatal and external forces from repetitive sleep positioning may cause these self-limiting skull shape abnormalities. In 1992, the American Academy of Pediatrics recommended that infants sleep on their backs to reduce the risk of sudden infant death syndrome. An increased number of referrals for nonsynostotic posterior plagiocephaly has resulted because of this “Back to Sleep” campaign and the increased awareness of cranial vault deformities within the pediatric community.
Proper identification of cranial vault deformities resulting from nonsynostotic causes (ie, positional plagiocephaly) and those associated with a true absence of a cranial vault suture (ie, craniosynostosis) is critical for diagnosis and appropriate treatment. The importance of a correct diagnosis is underscored by the dramatically different management approaches for each infant. Craniosynostosis requires surgical treatment, whereas benign positional skull molding requires conservative nonsurgical management using custom-made cranial orthotic devices and is not associated with neurologic or developmental sequelae.
A careful history and clinical examination are the primary basis for diagnosis of craniosynostosis, because each form produces a unique head shape. Because the condition is an intrauterine event, parents often report that the deformity was noted immediately after birth and without subsequent improvement. By contrast, cases of positional plagiocephaly have well-rounded head shape at birth, with asymmetry developing 3 to 6 months later (suggesting postnatal deformation of a flexible cranial vault). A consistent combination of arrested growth in one region and compensatory increase in others may explain unilateral sutural involvement causing a bilateral deformity or posterior suture fusion producing an anteroposterior (A-P) dimension deformity. An examiner familiar with these aberrant growth patterns and the characteristic dysmorphologies specific to each type of synostosis should consistently establish an accurate clinical diagnosis.
Craniosynostosis is primarily diagnosed by careful clinical examination but must be confirmed radiographically. Following clinical evaluation by a craniofacial team, a complete skull series of plain radiographs is obtained to evaluate the cranial vault sutures (usually not needed for the simple suture synostosis). Standard views of the cranial vault are often adequate to establish the absence of a cranial vault suture and confirm the diagnosis. For cases in which all the sutures cannot be adequately visualized using plain films, additional imaging using computed tomography (CT) is indicated. In addition to confirming the diagnosis, high-quality craniofacial CT scans provide more detailed 3-dimensional morphologic information, which is useful during the surgical planning phase. One-millimeter axial and coronal cuts with overlap and 3-dimensional reconstructions of the cranial vault, cranial base, orbits, and maxillofacial skeleton are recommended. Precise patient positioning is important in obtaining a diagnostic scan, and this may require sedation or general anesthesia.
Although technological advancements have dramatically increased the accuracy of standard radiographic studies and CT imaging, incorrect and outdated terminology is still occasionally used in describing cranial vault sutures. At some medical centers, radiologists and surgeons continue to use terms such as fibrous synostosis, suture dysfunction, and impending synostosis when describing cranial vault sutures that otherwise appear open. It is known that craniosynostosis is most often an intrauterine event that is usually recognized at birth. Therefore, a patent suture on a diagnostic-quality radiographic study negates the diagnosis of craniosynostosis. Although the postnatal fusion of cranial vault sutures has been described in the scientific literature, it is considered an extremely rare event that is specifically related to certain clinical and syndromic conditions. Some terms not consistent with contemporary knowledge about the condition may be confusing to parents and health care providers, perhaps resulting in inappropriate management.
Treatment of nonsyndromic craniosynostosis
General Considerations
The 2 primary objectives in the contemporary surgical management of nonsyndromic craniosynostosis are release of the involved (ie, fused) suture to allow brain growth in an unrestricted fashion and reconstruction of all dysmorphic skeletal components to achieve a more anatomically correct shape. The best results are often seen with collaboration between a well-organized craniofacial team experienced in managing these issues. Involvement of several other pediatric subspecialists, including geneticists, ophthalmologists, social workers, pediatric anesthesiologists, and pediatric intensivists is mandatory for a successful team.
Early surgical techniques used to treat craniosynostosis involved only the removal of involved sutures via a strip craniectomy. Generally, these limited craniectomy procedures would be performed by a neurosurgeon working independently. The theory behind this approach was that release of the suture would allow unrestricted brain growth and that the expanding brain would adequately recontour the bones without the need for formal craniofacial reconstruction. Although this approach does allow for cerebral decompression, a dysmorphic skull does not reshape itself, even in the presence of an expanding brain, and the result is a residual bony deformity.
Modern surgical management of craniosynostosis involves release of the involved sutures with a formal craniotomy performed by a neurosurgeon. Reconstruction requires the removal, dismantling, and reassembly of all dysmorphic skeletal components into a more appropriate anatomic position. Depending on which specific cranial vault suture is affected, the reconstruction may also involve the orbits. The exact surgical plan is formulated based on the extent of the skeletal deformity, the sutures involved, and the age of the patient at the time of diagnosis. In most cases of nonsyndromic single-suture craniosynostosis, one definitive surgical procedure is required to simultaneously release the suture and reshape the affected skull. This is more likely when the surgical procedure is performed during the first year of life and the patient has normal brain growth.
Timing of Surgery
Craniosynostosis diagnosed early does not represent a “surgical urgency,” because the risk of developing increased ICP is minimal during the early growth period (while the fontanelles are open). In most patients with single-suture abnormality, elevated ICP is not frequently encountered and the cranial vault dysmorphology can be safely addressed in the first year of life. The specific, ideal age at which to proceed with craniosynostosis repair is controversial, leading to a wide range of recommendations among pediatric craniofacial and neurologic surgeons. Some advocate an early repair performed at 3 to 6 months of age, whereas others postpone surgery until after age 9 to 11 months. There are theoretical advantages and potential negative consequences associated with each approach.
Some surgeons recommended early surgical correction (3–6 months of age), suggesting that early suture release allows natural brain growth to mold the developing cranium. Such an approach minimizes the surgical bony remodeling and offers the theoretical advantage; however, this method does not obviate the need for formal skeletal reconstruction to establish desired anatomic configuration. Bone segments in younger infants are more malleable, which permits easier reshaping, and the growing brain does provide rapid elimination of extradural dead space and support for the newly constructed bony framework.
Delaying the surgical procedure until approximately 9 to 11 months of age permits a greater proportion of the child’s cranial vault growth to occur before correction. This may translate into a more stable skeletal result with fewer postsurgical distortions related to subsequent growth. At this age, the bones are better ossified and harder, which results in less separation at suture lines (eg, frontozygomatic suture) and easier placement of rigid internal fixation devices.
When craniofacial reconstructive surgery is performed during infancy, complete healing of fairly large (1.5 cm) bony defects occurs without the need for additional bone grafting. This is due to the combined bony healing capacity of the pericranium and the dura, which is highly osteogenic during infancy. Most residual cranial vault defects heal completely when the surgery is performed during the first 2 years of life. When cranial vault surgery is undertaken between 2 and 4 years of life, complete healing of residual full-thickness defects is less predictable, and immediate grafting or a secondary procedure for repair may be indicated. After age 4 years, it is unlikely that even small full-thickness cranial vault defects would resolve without deliberate reconstruction or grafting at the time of the initial surgery.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


