Virchow, in 1851, coined the term craniosynostosis and formulated the classic theory known as Virchow’s law. This states that premature fusion of a cranial vault suture (craniosynostosis) inhibits skull growth perpendicular to the fused suture and results in a compensatory growth at the open sutures. The general direction of growth after synostosis is parallel to the fused suture.
The term craniofacial dysostosis (CFD) is used in a general way to describe syndromic forms of craniosynostosis. These disorders are characterized by sutural involvement that not only includes the cranial vault but also extends into the skull base and midfacial skeletal structures. In the past, CFD syndromes have been described by Carpenter, Apert, Crouzon, Saethre-Chotzen, and Pfeiffer. Although the cranial vault and cranial base are thought to be the regions of primary involvement, there is generally significant impact on midfacial growth and development. In addition to cranial vault dysmorphology, individuals with these inherited conditions exhibit a characteristic but variable “total midface” deficiency that is syndrome specific and must be addressed as part of the staged reconstructive approach. Advances in molecular genetics now offer a more accurate understanding of the basic biology of these syndromes.
GENETIC ASPECTS
FGFR-related CFD syndromes include: fibroblast growth factor receptor (FGFR)1-related craniosynostosis (Pfeiffer syndrome types 1, 2, and 3) ; FGFR2-related craniosynostosis (Apert syndrome, Beare-Stevenson syndrome, Crouzon syndrome, FGFR2-related isolated coronal synostosis, Jackson-Weiss syndrome, Pfeiffer syndrome types 1, 2, and 3); and FGFR3-related craniosynostosis (Crouzon syndrome with acanthosis nigricans, FGFR3-related isolated coronal synostosis, Muenke syndrome).
The eight disorders considered as part of the FGFR -related craniosynostosis spectrum are Pfeiffer syndrome, Aperts syndrome, Crouzon syndrome, Beare-Stevenson syndrome, FGFR2 -related isolated coronal synostosis, Jackson-Weiss syndrome, Crouzon syndrome with acanthosis nigricans, and Muenke syndrome. All but Muenke syndrome and FGFR2 -related isolated coronal synostosis generally have bicoronal synostosis or cloverleaf skull anomaly.
The diagnosis of Muenke syndrome ( FGFR3 -related coronal synostosis) is based on the identification of a disease-causing mutation in the FGFR3 gene. The diagnosis of FGFR2 -related isolated coronal synostosis is based on the identification of a disease-causing mutation in the FGFR2 gene . The diagnosis of the other six FGFR -related craniosynostoses is based on clinical findings; however, molecular genetic testing of the FGFR1, FGFR2, and FGFR3 genes may be helpful in establishing the diagnosis of these syndromes in questionable cases.
FGFR -related craniosynostosis is inherited in an autosomal dominant manner. Affected individuals have a 50% chance of passing the mutant gene to each child. Prenatal testing is available; however, its use is limited by poor predictive value.
Molecular testing is necessary to establish the diagnosis for two of the disorders, Muenke syndrome and FGFR2 -related isolated coronal synostosis . Individuals with Muenke syndrome may have unilateral coronal synostosis or megalencephaly without craniosynostosis. The accurate diagnosis depends upon the identification of a disease-causing mutation in the FGFR3 gene . FGFR2 -related isolated coronal synostosis is characterized only by unicoronal or bicoronal craniosynostosis. The accurate diagnosis depends upon the identification of a disease-causing mutation in the FGFR2 gene .
FUNCTIONAL CONSIDERATIONS
RESTRICTED BRAIN GROWTH AND INTRACRANIAL PRESSURE
If the rapid brain growth which normally occurs during infancy is to proceed unhindered, the cranial vault and skull base sutures must expand during phases of rapid growth, resulting in marginal ossification. In craniosynostosis, premature fusion of the suture(s) causes limited and abnormal skeletal expansion in the presence of continued brain growth. Depending on the number and location of prematurely fused sutures, the growth of the brain may be restricted. In addition, abnormal cranial vault and midfacial morphology occurs as determined by Virchow’s law. If surgical release of the affected suture(s) and reshaping of the involved skeleton to restore a more normal intracranial volume and configuration is not performed, decreased cognitive and behavioral function is likely to be the end result.
Elevated intracranial pressure (ICP) is the most serious functional problem associated with premature suture fusion. Radiographic findings that may suggest an elevated ICP include the “beaten-copper” appearance along the inner table of the cranial vault seen on a plain radiograph or the loss of brain cisternae as observed on a computed tomography (CT) scan. Although suggestive of increased ICP, these are considered soft findings.
Increased ICP is most likely to affect patients with great disparity between brain growth and intracranial capacity and is thought to occur in as many as 42% of untreated children in whom two or more sutures are affected. Unfortunately, there is not agreement on what levels of ICP are “normal” at any given age in infancy and early childhood.
The clinical signs and symptoms related to elevated ICP may have a slow onset and be difficult to recognize in the pediatric population. Although standardized CT scans allow for indirect measurement of intracranial volume, it is not yet possible to use these studies to make judgments as to who requires craniotomy for brain decompression. Comprehensive neurologic and ophthalmologic evaluation are critical components of the data gathering required to formulate definitive treatment plans in patients with one of the CFD syndromes.
VISION
Untreated craniosynostosis with elevated ICP may cause papilledema and eventual optic nerve atrophy, resulting in partial or complete blindness. If the orbits are shallow (exorbitism) and the eyes are proptotic (exophthalmus), as occurs in the CFD syndromes, the cornea may be exposed and abrasions or ulcerations may occur. An eyeball extending outside of a shallow orbit is also a risk for trauma. If the orbits are extremely shallow, herniation of the globe itself may occur, necessitating emergency reduction followed by tarsorrhaphies or urgent orbital decompression.
Some forms of CFD (e.g., Apert syndrome) result in a degree of orbital hypertelorism, which may compromise visual acuity and restrict binocular vision. Divergent or convergent nonparalytic strabismus or exotropia occurs frequently and should be considered during the diagnostic evaluation. This may be the result of congenital anomalies of the extraocular muscles themselves. Paralytic or nonparalytic unilateral or bilateral upper eyelid ptosis also occurs with greater frequency than what is encountered in the general population.
HYDROCEPHALUS
Hydrocephalus affects as many as 10% of patients with a CFD syndrome. The risk of intracranial hypertension is greatest in Crouzon syndrome. Even if every medical complication is managed promptly, a proportion of affected children will develop cognitive delay and neurologic problems. Although the cause is often not clear, hydrocephalus may be secondary to a generalized cranial base stenosis with constriction of all the cranial base foramina, which impacts the patient’s cerebral venous drainage and cerebrospinal fluid (CSF) flow dynamics. Hydrocephalus may be identified with the help of a CT scan or magnetic resonance imaging (MRI) to document progressively enlarging ventricles. Difficulty exists in interpreting ventricular findings as seen on a CT scan, especially when the skull and cranial base are brachycephalic. The skeletal dysmorphology seen in a child with cranial dysmorphology related to craniosynostosis (e.g., bicoronal synostosis) may translate into an abnormal ventricular shape, which is not necessarily related to abnormal CSF flow. Serial imaging with clinical correlation and experienced neurologic judgment is required in making these assessments.
EFFECTS OF MIDFACE DEFICIENCY ON AIRWAY
All newborn infants are obligate nasal breathers. Many infants born with a CFD syndrome have moderate to severe hypoplasia of the midface as a component of their malformation. They will have diminished nasal and nasopharyngeal spaces with resulting increased nasal airway resistance (obstruction). The affected child is thus forced to breath through the mouth. For a newborn infant to ingest food through the mouth requires sucking from a nipple to achieve negative pressure and an intact swallowing mechanism. The neonate with severe midface hypoplasia will experience diminished nasal airflow and be unable to accomplish this task and breathe through the nose at the same time. Complicating this clinical picture may be an elongated and ptotic palate (e.g., Apert syndrome) and enlarged tonsils and adenoids. The compromised infant expends significant energy respiring, and this may push the child into a catabolic state (negative nitrogen balance). Failure to thrive results unless either nasogastric tube feeding is instituted or a feeding gastrostomy is placed. Evaluation by a pediatrician, pediatric otolaryngologist, and feeding specialist with craniofacial experience can help distinguish minor feeding difficulties from those requiring more aggressive treatment.
Sleep apnea of either central, obstructive, or mixed origin may also be present. If the apnea is found to be secondary to upper airway obstruction based on a formal sleep study, a tracheostomy may be indicated. In specific situations, “early” midface advancement maybe carried out to improve the airway allowing for tracheostomy decannulation. Central apnea may occur from poorly treated intracranial hypertension and other contributing factors. If so, the condition may improve by reducing the ICP through brain decompression. This is accomplished with cranio-orbital or posterior cranial vault expansion.
FUNCTIONAL CONSIDERATIONS
RESTRICTED BRAIN GROWTH AND INTRACRANIAL PRESSURE
If the rapid brain growth which normally occurs during infancy is to proceed unhindered, the cranial vault and skull base sutures must expand during phases of rapid growth, resulting in marginal ossification. In craniosynostosis, premature fusion of the suture(s) causes limited and abnormal skeletal expansion in the presence of continued brain growth. Depending on the number and location of prematurely fused sutures, the growth of the brain may be restricted. In addition, abnormal cranial vault and midfacial morphology occurs as determined by Virchow’s law. If surgical release of the affected suture(s) and reshaping of the involved skeleton to restore a more normal intracranial volume and configuration is not performed, decreased cognitive and behavioral function is likely to be the end result.
Elevated intracranial pressure (ICP) is the most serious functional problem associated with premature suture fusion. Radiographic findings that may suggest an elevated ICP include the “beaten-copper” appearance along the inner table of the cranial vault seen on a plain radiograph or the loss of brain cisternae as observed on a computed tomography (CT) scan. Although suggestive of increased ICP, these are considered soft findings.
Increased ICP is most likely to affect patients with great disparity between brain growth and intracranial capacity and is thought to occur in as many as 42% of untreated children in whom two or more sutures are affected. Unfortunately, there is not agreement on what levels of ICP are “normal” at any given age in infancy and early childhood.
The clinical signs and symptoms related to elevated ICP may have a slow onset and be difficult to recognize in the pediatric population. Although standardized CT scans allow for indirect measurement of intracranial volume, it is not yet possible to use these studies to make judgments as to who requires craniotomy for brain decompression. Comprehensive neurologic and ophthalmologic evaluation are critical components of the data gathering required to formulate definitive treatment plans in patients with one of the CFD syndromes.
VISION
Untreated craniosynostosis with elevated ICP may cause papilledema and eventual optic nerve atrophy, resulting in partial or complete blindness. If the orbits are shallow (exorbitism) and the eyes are proptotic (exophthalmus), as occurs in the CFD syndromes, the cornea may be exposed and abrasions or ulcerations may occur. An eyeball extending outside of a shallow orbit is also a risk for trauma. If the orbits are extremely shallow, herniation of the globe itself may occur, necessitating emergency reduction followed by tarsorrhaphies or urgent orbital decompression.
Some forms of CFD (e.g., Apert syndrome) result in a degree of orbital hypertelorism, which may compromise visual acuity and restrict binocular vision. Divergent or convergent nonparalytic strabismus or exotropia occurs frequently and should be considered during the diagnostic evaluation. This may be the result of congenital anomalies of the extraocular muscles themselves. Paralytic or nonparalytic unilateral or bilateral upper eyelid ptosis also occurs with greater frequency than what is encountered in the general population.
HYDROCEPHALUS
Hydrocephalus affects as many as 10% of patients with a CFD syndrome. The risk of intracranial hypertension is greatest in Crouzon syndrome. Even if every medical complication is managed promptly, a proportion of affected children will develop cognitive delay and neurologic problems. Although the cause is often not clear, hydrocephalus may be secondary to a generalized cranial base stenosis with constriction of all the cranial base foramina, which impacts the patient’s cerebral venous drainage and cerebrospinal fluid (CSF) flow dynamics. Hydrocephalus may be identified with the help of a CT scan or magnetic resonance imaging (MRI) to document progressively enlarging ventricles. Difficulty exists in interpreting ventricular findings as seen on a CT scan, especially when the skull and cranial base are brachycephalic. The skeletal dysmorphology seen in a child with cranial dysmorphology related to craniosynostosis (e.g., bicoronal synostosis) may translate into an abnormal ventricular shape, which is not necessarily related to abnormal CSF flow. Serial imaging with clinical correlation and experienced neurologic judgment is required in making these assessments.
EFFECTS OF MIDFACE DEFICIENCY ON AIRWAY
All newborn infants are obligate nasal breathers. Many infants born with a CFD syndrome have moderate to severe hypoplasia of the midface as a component of their malformation. They will have diminished nasal and nasopharyngeal spaces with resulting increased nasal airway resistance (obstruction). The affected child is thus forced to breath through the mouth. For a newborn infant to ingest food through the mouth requires sucking from a nipple to achieve negative pressure and an intact swallowing mechanism. The neonate with severe midface hypoplasia will experience diminished nasal airflow and be unable to accomplish this task and breathe through the nose at the same time. Complicating this clinical picture may be an elongated and ptotic palate (e.g., Apert syndrome) and enlarged tonsils and adenoids. The compromised infant expends significant energy respiring, and this may push the child into a catabolic state (negative nitrogen balance). Failure to thrive results unless either nasogastric tube feeding is instituted or a feeding gastrostomy is placed. Evaluation by a pediatrician, pediatric otolaryngologist, and feeding specialist with craniofacial experience can help distinguish minor feeding difficulties from those requiring more aggressive treatment.
Sleep apnea of either central, obstructive, or mixed origin may also be present. If the apnea is found to be secondary to upper airway obstruction based on a formal sleep study, a tracheostomy may be indicated. In specific situations, “early” midface advancement maybe carried out to improve the airway allowing for tracheostomy decannulation. Central apnea may occur from poorly treated intracranial hypertension and other contributing factors. If so, the condition may improve by reducing the ICP through brain decompression. This is accomplished with cranio-orbital or posterior cranial vault expansion.
DENTITION AND OCCLUSION
The incidence of dental and oral anomalies is higher among children with CFD syndromes than within the general population. In Apert syndrome in particular, the palate is high and constricted in width. The incidence of an isolated cleft palate in patients with Apert syndrome approaches 30%. Clefting of the secondary palate may be submucous, incomplete, or complete. Delayed dental eruption should also be expected. Confusion has arisen over whether the oral malformations and absence of teeth that are often characteristic of these conditions are a result of congenital or iatrogenic factors (e.g., injury to dental follicles associated with early midface surgery). The midfacial hypoplasia seen in the CFD syndromes often results in limited maxillary alveolar bone to house a full complement of teeth. The result is severe crowding, which often requires serial extractions to address it. An Angle’s Class III skeletal relationship in combination with an anterior open bite deformity is typical.
HEARING
Hearing deficits are more common among patients with the CFD syndromes than among the general population. In Crouzon syndrome, conductive hearing deficits are common, and atresia of the external auditory canals may also occur. Otitis media is more common in Apert syndrome, although the exact incidence is unknown. Middle ear disease may be related to the presence of a cleft palate that results in eustachian tube dysfunction. Congenital fixation of the stapedial footplate is also believed to be a frequent finding. The possibility of significant hearing loss is paramount in importance and should not be overlooked because of preoccupation with other more easily appreciated craniofacial findings.
EXTREMITY ANOMALIES
Apert syndrome results in joint fusion and bony and soft tissue syndactylism of the digits of all four limbs. These Apert-associated extremity deformities are often symmetrical. Partial or complete fusion of the shoulder, elbow, or other joints is common. Broad thumbs, broad great toes, and partial soft tissue syndactyly of the hands may be seen in Pfeiffer syndrome, but these are variable features. Preaxial polysyndactyly of the feet may also be seen in Carpenter syndrome.
MORPHOLOGIC CONSIDERATIONS
GENERAL
Examination of the patient’s entire craniofacial region should be meticulous and systematic. The skeleton and soft tissues are assessed in a standard way to identify all normal and abnormal anatomy. Specific findings tend to occur in particular malformations, but each patient is unique. The achievement of symmetry, normal proportions, and the reconstruction of specific aesthetic units is essential to forming an unobtrusive face in a child born with one of the CFD syndromes.
FRONTOFOREHEAD AESTHETIC UNIT
The frontoforehead region is dysmorphic in an infant with CFD. Establishing the normal position of the forehead is critical to overall facial symmetry and balance. The forehead maybe considered as two separate aesthetic components: the supraorbital ridge-lateral orbital rim region and the superior forehead. The supraorbital ridge-lateral orbital rim region includes the nasofrontal process and supraorbital rim extending inferiorly down each frontozygomatic suture toward the infraorbital rim and posteriorly along each temporoparietal region. The shape and position of the supraorbital ridge-lateral orbital rim region is a key element of upper facial aesthetics. In a normal forehead, at the level of the nasofrontal suture, an angle ranging form 90° to 110° is formed by the supraorbital ridge and the nasal bones when viewed in profile. Additionally the eyebrows, overlying the supraorbital ridge, should be anterior to the cornea. When the supraorbital ridge is viewed from above, the rim should arc posteriorly to achieve a gentle 90° angle at the temporal fossa with a center point of the arc at the level of each frontozygomatic suture. The superior forehead component, about 1.0 to 1.5 cm up form the supraorbital rim, should have a gentle posterior curve of about 60°, leveling out at the coronal suture region when seen in profile.
POSTERIOR CRANIAL VAULT AESTHETIC UNIT
Symmetry, form, and adequate intracranial volume of the posterior cranial vault are closely linked. Posterior cranial vault flattening may result from either a unilateral or bilateral lambdoidal synostosis, which is rare; previous craniectomy with reossification in a dysmorphic flat shape, which is frequent; or postural molding because of repetitive supine sleep positioning. A short anterior-posterior (AP) cephalic length may be misinterpreted as an anterior cranial vault (forehead) problem when the occipitoparietal (posterior) skull represents the primary region of the deformity. Careful examination of the entire cranial vault is essential to defining the dysmorphic region so that, when indicated, appropriate cranial vault expansion may be carried out.
ORBITO-NASO-ZYGOMATIC AESTHETIC UNIT
In the CFD syndromes, the orbito-naso-zygomatic regional deformity is a reflection of the cranial base malformation. For example, in Crouzon syndrome when bilateral coronal suture synostosis is combined with skull base and midfacial deficiency, the orbito-naso-zygomatic region will be dysmorphic and consistent with a short (AP) and wide (transverse) anterior cranial base. In Apert syndrome, the nasal bones, orbits, and zygomas, like the anterior cranial base, are transversely wide from anterolateral bulging of the temporal lobes of the brain and horizontally short (retruded) resulting in a shallow hyperteloric “reverse curved” upper midface (zygomas, orbits, and nose). Surgically advancing the midface without simultaneously addressing the increased transverse width and “reverse curve” will not adequately correct the dysmorphology.
MAXILLARY-NASAL BASE AESTHETIC UNIT
In the CFD patient with midface deficiency, the upper anterior face is vertically short (nasion to maxillary incisor), and there is a lack of horizontal (AP) projection of the midface. These findings may be confirmed through cephalometric analysis that indicates an SNA angle below the mean value and a short upper anterior facial height (nasion to anterior nasal spine). The width of the maxilla in the dentoalveolar region is generally constricted with a high arched palate. To normalize the maxillary-nasal base region, multidirectional surgical expansion and reshaping are generally required. The abnormal maxillary lip-to-tooth relationship and occlusion are improved through Le Fort I segmental osteotomies and orthodontic treatment as part of the staged reconstruction. The mandible and chin are frequently secondarily deformed and benefit from surgical repositioning as part of the orthognathic correction.
SURGICAL MANAGEMENT
HISTORICAL PERSPECTIVES
As early as 1890, Lannelongue and then Lane reported a surgical approach to the treatment of craniosynostosis. Lannelongue’s aim was to remove the fused suture (strip craniectomy) in the hope of controlling the problem of brain compression within a congenitally small cranial vault. By the turn of the century, Harvey Cushing, the most prominent neurosurgeon of his day, suggested that surgical intervention for the problem of craniosynostosis was misdirected, and more attention should instead be given to the schooling of these children. Schillito disagreed with Cushing and enthusiastically supported the concept of surgical intervention to improve the outlook for these children. He believed that the linear “strip” craniectomy of the fused sutures would “release” the skull and allow the cranium to reshape itself and continue to grow in a normal and symmetrical fashion. The strip craniectomy procedures were supposed to allow for a new suture line at the site of the previous synostosis. With the realization that this goal was not biologically feasible, attempts were made to remove portions of the cranial vault surgically and to either leave large open areas or to use the removed segments as free grafts to refashion the cranial vault shape. Problems with these methods included uncontrolled postoperative skull molding, reossification into dysmorphic configurations, and the occurrence of large residual skull defects.
The concept of a simultaneous release of the fused suture combined with more meticulous cranial vault reshaping carried out in infants was initially suggested by Rougerie et al and then refined by Hoffman and Mohr in 1976 for children born with unilateral coronal synostosis. Whitaker and others proposed a more formal anterior cranial vault and orbital reshaping procedure for unilateral coronal synostosis in 1977. Marchac and Renier published their experience with a “floating forehead” technique for simultaneous unilateral coronal and bilateral coronal suture release and forehead and upper orbital osteotomies with advancement to manage craniosynostosis in infancy. Unfortunately, the “floating forehead” technique resulted in unpredictable reossification of the open cranial vault areas. In addition, bitemporal constrictions or bulging and vertex of the skull concavities or bulging were frequent occurrences as reossification occurred. In any case, the hoped for midface growth did not materialize.
In the 1950s, Gillies and Harrison reported experience with an extracranial Le Fort III “type” of osteotomy to improve the anterior projection of the midface in an adult with Crouzon syndrome. The initial Gillies procedure was actually carried out in 1942. He mobilized the midface by a variety of osteotomies preformed through skin excisions directly over each osteotomy site. The midface was mobilized, advanced, and intermaxillary fixation (IMF) applied. Following removal of the IMF (2 weeks after the operation), the metal cast-cap (dental) splint was attached to a plaster headcap, which was maintained for 3 weeks. There is no evidence that Gillies used any bone grafts in the surgical gaps created. The early enthusiasm for this technique later turned to discouragement when the patient’s facial skeleton relapsed to its preoperative status. There was relapse—both at the maxillary incisor level and of the eye proptosis.
Dr. Tessier was aware of Gillies’ previous work and the accompanying difficulties. In 1967 Tessier described a new (intracranial-cranial base) approach to the management of Crouzon syndrome. This work was first presented in France at a meeting in Montpelier in 1966 and the following year at the International Plastic Surgery Meeting in Rome. Tessier’s landmark presentations and publications were the beginning of modern craniofacial surgery. To overcome the earlier problems encountered by Gillies and Harrison, Tessier developed an innovative basic surgical approach that included new locations for the Le Fort III osteotomy, a combined intracranial-extracranial (cranial base) approach, the use of a coronal (skin) incision to expose the upper facial bones, and the use of a fresh autogenous bone graft. He also applied an external fixation device to help maintain bony stability until healing had occurred.
In 1971 Tessier described a single-stage frontofacial advancement in which the fronto-orbital bandeau was advanced as a separate element in conjunction with the Le Fort III complex below and the frontal bones above. Seven years later, Ortiz-Monasterio et al refined the “monobloc” osteotomy to advance the orbits and midface as one unit, combined with frontal bone (anterior cranial vault) repositioning to correct the upper face and midface deformity of Crouzon syndrome. In 1979 Van der Meulen described the “medial fasciotomy” for the correction of midline facial clefting. Van der Meulen split the monobloc osteotomy vertically in the midline, removed central nasal and ethmoid bone, and then moved the two halves of the facial skeleton together for correction of the orbital hypertelorism. To correct the midface dysplasia and associated orbital hypertelorism in Apert syndrome, Tessier refined the vertical splitting and reshaping of the midline split monobloc segments, thus correcting the midface deformity in three dimensions in a procedure now known as facial bipartition . Wolfe et al, Kowamoto, and Posnick et al have independently documented the advantages of Tessier’s facial bipartition technique for the correction of the upper face and midface deformities of Apert’s syndrome.
The widespread use of autogenous cranial bone grafting has virtually eliminated the need for rib and hip grafts when bone replacement is required in cranio-orbito-zygomatic procedures. This represents another of Tessier’s contributions to craniofacial surgery.
In 1968 Luhr introduced the use of small metal (Vitallium) plates and screws to stabilize maxillofacial fractures and then osteotomies. In current practice, the use of internal miniplate and microplate and screw (titanium) fixation (of various sizes) is the preferred form of fixation when stability and three-dimensional craniomaxillofacial reconstruction of multiple osteotomized bone segments and grafts are required. In infants and young children, for the stabilization of non–load-bearing osteotomy segments (i.e., cranial vault), stabilization with resorbable plates and screws is frequently used.
GENERAL CONSIDERATIONS
Philosophy Regarding Timing of Intervention
In considering the timing and type of intervention, the experienced surgeon will take several biologic realities into account: the natural course of the malformation (i.e., is the dysmorphology associated with Crouzon syndrome progressively worsening or only a nonprogressive craniofacial deformity?); the tendency toward growth restriction of operated bones (aesthetic units) that have not yet reached maturity (i.e., we know that operating on a cleft palate in infancy will cause scarring and later result in maxillary hypoplasia in a significant percentage of individuals); and the relationship between the underlying growing viscera (i.e., brain or eyes) and the congenitally affected and surgically altered skeleton (i.e., in a Crouzon syndrome, if the cranial vault is not surgically expanded to decompress the brain by 1 year of life, is brain compression likely to occur?).
In attempting to limit impairment and also achieve long-term preferred facial aesthetics and head and neck function, an essential question the surgeon must ask is, “ during the course of craniofacial development, does the operated-on facial skeleton of the craniofacial dysostosis child tend to grow abnormally, resulting in further distortions and dysmorphology, or are the initial positive skeletal changes achieved (at operation) maintained during ongoing growth ?” Unfortunately, the proposed theory that craniofacial procedures carried out early in infancy will “unlock growth” has not been documented through the scientific method.
Incision Placement
For exposure of the craniofacial skeleton above the Le Fort I level, the approach used is the coronal (skin) incision. This allows for a relatively camouflaged access to the anterior and posterior cranial vault, orbits, nasal dorsum, zygomas, upper maxilla, pterygoid fossa, and temporomandibular joints. For added cosmetic advantage, placement of the coronal incision more posterior on the scalp and with postauricular rather than preauricular extensions is useful. When exposure of the maxilla at the Le Fort I level is required, a circumvestibular maxillary intraoral incision is used. Unless complications occur that warrant unusual exposure, no other incisions are required for managing any aspect of the CFD patient’s reconstruction. These incisions (coronal [scalp] and maxillary [circumvestibular]) may be reopened as needed to further complete the individual’s staged reconstruction.
Management of the Cranial Fossa Dead Space and Communication Across the Skull Base After Total Midface Advancement
Cranial reshaping in the CFD patient provides space for the compressed brain to expand. Unfortunately, after anterior cranial vault expansion and monobloc advancement, an immediate extradural (retrofrontal) dead space is combined with the osteotomy-created gap across the skull base (connecting the anterior cranial fossa and the nasal cavity). This combination of factors may complicate the postoperative recovery (e.g., CSF leakage, infection, bone loss, fistula formation). After frontofacial advancement, the nasal cavity-cranial fossa communication is managed by: being gentle to the tissues; good hemostasis; the effective repair of any dural tears (dural grafting as needed); the complete separation of dural and nasal mucosal tissue planes by interposing a combination of bone grafts, tissue sealants, and flaps; avoidance of pressure gradients across the opening while the nasal mucosa is healing; and prevention of “overshunting” or “undershunting” (when a shunt is in place.)
The preferred way to manage the retrofrontal (lobes of the brain) dead space and the gap across the skull base osteotomy site (separating the cranial fossa and nasal cavities) following frontofacial advancement remains controversial, but all agree that it is a critical aspect of the reconstruction.
In the CFD patient, relatively rapid filling of the surgically expanded intracranial volume (6 to 8 weeks) by the previously compressed frontal lobes of the brain has been documented after cranio-orbital expansion in infants. It has also been shown to occur after frontofacial advancement in children and young adults when the volume increase remains in a physiologic range. These observations support the conservative management of the retrofrontal dead space in younger patients. More gradual and less complete filling of the space is thought to occur in older children and adults. If so, this may be particularly troublesome when the anterior cranial fossa dead space communicates directly with the nasal cavity (i.e., monobloc advancement, facial bipartition, intracranial Le Fort III) across the (open-gap) skull base interface. When feasible, closing off (sealing) the nasal cavity from the cranial fossa across the skull base osteotomy at the time of operation is preferred. Insertion of a pericranial flap or other fillers can help to separate the cavities. The use of fibrin glue to seal the anterior cranial base provides a temporary separation between the cavities, allowing time for the reepithelialization (healing) of the torn nasal mucosa. To reconstruct the defect across the skull base (gap), bone grafts of various types have also been used. Until the torn nasal mucosa heals, potential communication between the nasal cavity and cranial fossa may result in the transfer of air, fluid, bacteria, and nasocranial fistula formation. To facilitate nasal mucosa healing and limit a pressure gradient across the communication, postoperative endotracheal intubation may be extended for 3 to 5 days, and/or bilateral nasopharyngeal airways may be placed after extubation. The avoidance of positive pressure ventilation, enforcement of sinus precautions, and restriction of nose blowing further limit reflux of air, fluid, and bacteria (nose to cranial fossa) during the early postoperative period. When anterior cranial vault procedures are performed and aerated frontal sinuses are present, management is by either cranialization or obliteration.
Aside from a learning curve in mastering the technical skills of completing the monobloc osteotomies and disimpaction, the surgical morbidity from these procedures primarily results from a combination of the anticipated retrofrontal dead space, unavoidable tears in the nasal mucosa, and management of nasocranial communication across the skull base gap with the potential for fluid, air, and bacteria contamination. Two benchmark studies clarify these issues.
Posnick et al studied the issue of retrofrontal dead space, the communication across the skull base, nasal mucosal lacerations, and associated morbidity in a consecutive series of patients (N = 23) undergoing either monobloc or facial bipartition osteotomies combined with cranial expansion by a single surgeon (JCP) during a 4-year time frame (1987 to 1991). The extradural (retrofrontal) dead space was measured from consistent CT scan images at specific postoperative intervals (immediate, 6 to 8 weeks, and 1 year). The study confirmed the presence of an immediate retrofrontal dead space, which generally filled in with the expanding brain and/or dura by 6 to 8 weeks after surgery. Specific intraoperative measures were taken by the surgeon to close (seal) the nasofrontal communication using flaps, fibrin glue, and Gelfoam. Precautions to prevent a pressure gradient across the communication (repair of dural tears, sinus precautions, and nasal stinting) were meticulously adhered to with the objective of providing time for nasal mucosal healing. The infection rate in this study group was limited to 2 of 23 patients or 9%. In both patients who developed infection, a retrofrontal (extradural) fluid collection with drainage across the residual nasofrontal communication occurred. Both patients healed without major sequelae, but did require further reconstruction of resorbed portions of the cranial vault.
Bradley et al recently completed a single-center retrospective chart review study comparing differences in morbidity in a series of CFD patients undergoing combined cranial vault expansion and monobloc advancement for correction of the upper and middle face deformities and/or hypoplasia. They describe three different sequential treatment groups over a period of 23 years. Group I patients (1979 to 1989; N = 12) underwent a monobloc advancement without any special attention to the retrofrontal dead space or the communication through the skull base between the anterior cranial fossa and the nasal cavity. Group II (1989 to 1995; N = 11) patients underwent varied attempts at closure of the skull base gap with pericranial flaps and fibrin glue. Group III (1995 to 2002; N = 24) patients underwent monobloc osteotomies and disimpaction but without any immediate advancement. An internal distraction device was placed across the osteotomized zygoma on each side. After 7 days, the monobloc and forehead advancement was initiated at 1 mm per day for approximately 2 to 4 weeks. The infection rate for group III patients was significantly lower (2 of 24 patients or 8%) than for those in group I and II. Neither of the two infections in group III resulted in bone loss. Not surprisingly, the group I patients had the greatest morbidity.
Advancement of the monobloc osteotomy in the CFD patient using the D.O. technique described by Bradley et al is likely to facilitate early nasal mucosa healing and thereby limit communication of fluid, air, and bacteria across the surgically created skull base gap. By allowing a 1-week delay in the actual advancement, the nasal mucosa may achieve sufficient healing to explain a drop in the infection rate from the 9% described by Posnick et al to the 8% described by Bradley’s group III patients.
Bradley et al also describe a greater advancement in their group III (D.O. treatment) patients compared with their group I and II patients. Confounding variables in their study may explain these differences, including greater experience and improved surgical technique, by the surgeons in the later years of their study (group III patients) and the effect of comorbidities (e.g., high rate of infection in group I and group II patients that likely increased relapse and limited long-term advancement). More importantly, no correlation is shown by Bradley et al between a number of millimeters of advancement of the frontal facial osteotomies in the CFD patient and either greater functional gains or enhanced facial aesthetics. In fact, with a monobloc (frontofacial) osteotomy as much aesthetic damage is done by overadvancement (enophthalmos) as by underadvancement (residual eye proptosis). In addition, the achievement of a “normal” occlusion is rarely a treatment objective at the time of a monobloc advancement. Accomplishing an ideal occlusion without creating enophthalmos requires a separate Le Fort I osteotomy to differentially advance the maxilla, often combined with maxillary segmentation and mandibular (sagittal split) osteotomies. To achieve the most favorable facial balance for the CFD patient, an experienced clinician’s aesthetic sense of the preferred morphology and focused technical expertise to alter the skeleton intraoperatively is essential. Several key technical aspects include:
- 1.
The ability to remove, segment, and then reshape and stabilize (plates and screws) the anterior cranial vault
- 2.
The ability to separate the orbits and midface as a unit (monobloc) from the skull base
- 3.
The ability to further segment the monobloc (at the upper orbits) and reconstruct (with cranial grafts) as needed
- 4.
The ability to separate the monobloc into halves (facial bipartition) and then alter the two facial halves to achieve the most favorable morphology. This often requires simultaneously increasing the maxillary transverse width and decreasing the upper face width to correct hypertelorism of the orbits, zygomas, nose, and bitemporal regions (e.g., Apert syndrome). Facial bipartition also provides the ability to correct the transverse facial arc of rotation deformities. For example, changing the Apert syndrome patients’ concave facial arc of rotation towards a normal convexity is an essential aspect of the reconstruction.
Any potential advantage of limiting morbidity as a result of infection across the skull base with the D.O. technique should be considered in light of limitations to achieve the above-mentioned key technical and aesthetic aspects. Added morbidity with the D.O. technique as a result of pin tract infection, scarring, and/or loosening requiring reapplication; the need for device removal; and dependence on a patient’s, family’s, and clinician’s continued commitment to staying the course for necessary outpatient D.O. procedures or adjustments to achieve an acceptable result must also be factored into the decision-making process.
When a CFD patient is to undergo intracranial volume expansion as part of the craniofacial procedure and they also require hydrocephalus management, the potential for morbidity increases. Complications may arise from excessive CSF drainage (overshunting). With “overshunting,” there is decreased brain volume to fill any surgically created retrofrontal dead space. Frontofacial advancement and/or cranial vault expansion procedures should be carefully staged with ventriculoperitoneal (VP) shunting procedures. We believe that the presence or absence of a VP shunt is not in itself a major risk factor in the success of a frontofacial advancement procedure. An important aspect is the satisfactory physiologic function of the ventricular system. Ultimately the decision regarding the need for and sequencing of shunting is based on the patient’s neurologic findings and the neurosurgeon’s judgment. In a patient with a VP shunt in place before the surgery, an experienced neurosurgical evaluation, including CT scanning of the ventricular system, is carried out to confirm physiologic shunt function.
Soft Tissue Management
A layered closure of the coronal (scalp) incision (galea and skin) optimizes healing and limits scar widening. Resuspension of the midface periosteum to the temporalis fascia may facilitate redraping of the soft tissues. Each lateral canthus should be reattached in a superior-posterior direction to the newly repositioned lateral orbital rim. The use of chromic gut on the skin in children may be used to obviate the need for postoperative suture or staple removal.
CROUZON SYNDROME
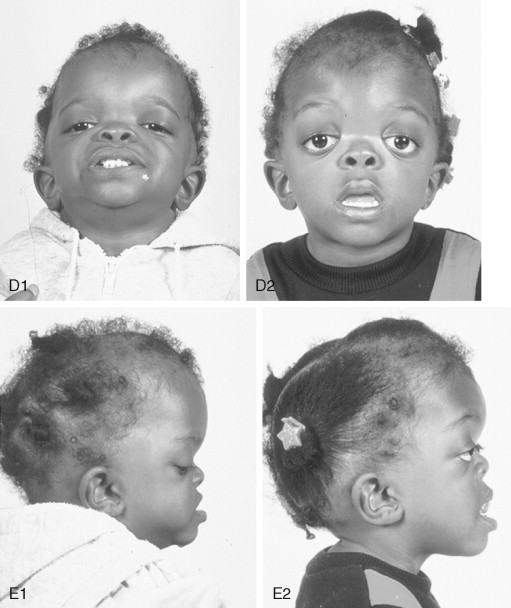
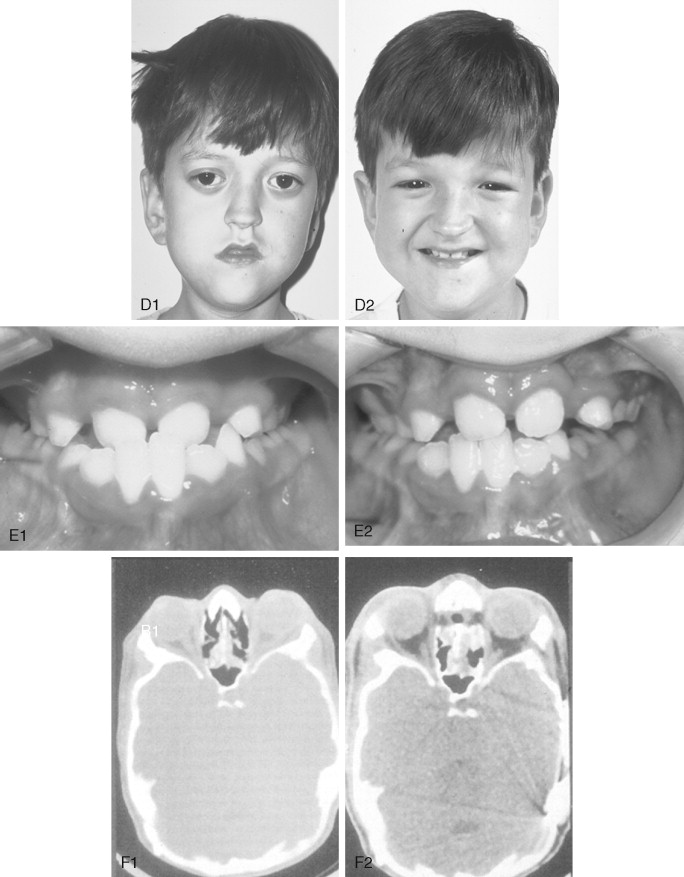
Crouzon syndrome is a frequent form of CFD. It is characterized by multiple anomalies of the craniofacial skeleton with an autosomal dominant inheritance pattern. Its manifestations are generally less severe than those of Apert syndrome, and there are no malformations of the extremities. Typically the cranial vault presentation is premature synostosis of both coronal sutures with a resultant brachycephalic shape to the skull. Cranial vault suture involvement other than coronal may include sagittal, metopic, or lambdoidal, either in isolation or in any combination. The cranial base and upper face sutures are variably involved, resulting in a degree of midface hypoplasia with an Angle Class III malocclusion. The orbits are hypoplastic, resulting in a degree of proptosis with additional orbital dystopia that may produce a mild orbital hypertelorism and flatness to the (transverse) arc of rotation of the midface.
By clinical examination alone, it may be difficult to accurately separate nonsyndromic bilateral coronal synostosis from Crouzon syndrome, with its expected midface deficiency. It is now known that Crouzon syndrome is caused by multiple mutations in the FGFR2 gene, some of which are identical to those seen in Pfeiffer and Jackson-Weiss syndromes. It is likely that in the past many of these individuals have been misdiagnosed as having Crouzon syndrome. The 5% of individuals with Crouzon syndrome who have acanthosis nigricans (pigmentary changes in the skin fold regions) are said to have Crouzon syndrome with acanthosis nigricans. It can be present in the neonatal period or appear later. Crouzon syndrome with acanthosis nigricans has been described with a specific Ala391Glu mutation in FGFR3 .
STAGING OF RECONSTRUCTION FOR CROUZON SYNDROME
Primary Brain Decompression: Cranio-orbital Reshaping in Infancy
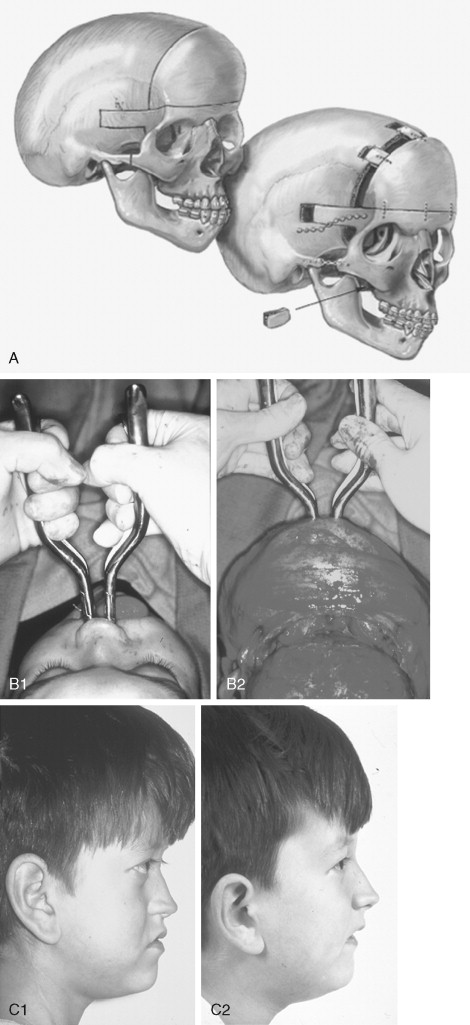
The initial treatment for any CFD syndrome generally requires cranial vault suture release and simultaneous anterior cranial vault and upper orbital osteotomies with reshaping and advancement for brain decompression ( Figure 45-1 ). In general, our preference is to carry this out when the child is 9 to 12 months of age unless clear signs of increased ICP are identified earlier in life. Reshaping of the upper three quarters of the orbital rims, supraorbital ridges, and tenon extensions is geared to decreasing the bitemporal and anterior cranial base width, with simultaneous horizontal advancement to increase the AP dimension. This also increases the depth of the upper orbits, with some improvement of eye proptosis. The overlying forehead is then reconstructed according to morphologic needs. A degree of overcorrection is considered at the level of the supraorbital ridge when the procedure is carried out in infancy. In our opinion, by allowing additional growth to occur before surgery (waiting until the child is 9 to 12 months old), the reconstructed cranial vault and upper orbital shape is better maintained with less need for repeat craniotomy procedures in early childhood. Preoperative evaluation and monitoring confirms that waiting until this age (9 to 12 months) will not risk compression of the underlying brain.
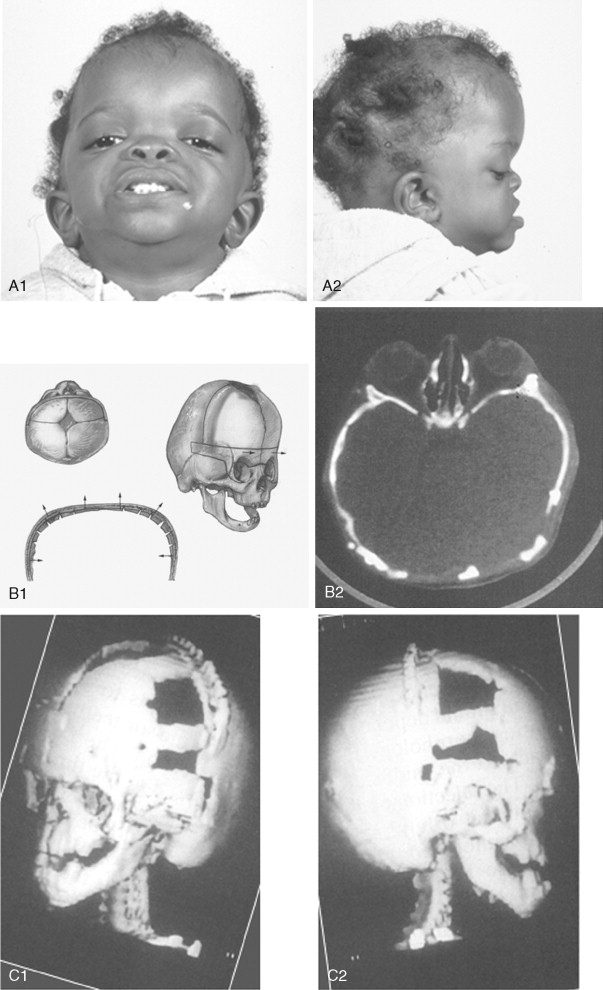
The goals at this stage are to provide increased space in the anterior cranial vault for the brain; to increase the orbital volume, which allows the eyes to be positioned more normally for better protection from exposure; and to improve the morphology of the forehead and upper orbits.
A postauricular coronal (scalp) incision is made, and the anterior flap is elevated along with the temporalis muscle in the subperiosteal plane. Bilateral circumferential periorbital dissection follows with detachment of the lateral canthi but with preservation of the medial canthi and nasolacrimal apparatus to the medial orbital walls. The subperiosteal dissection is continued down the lateral and infraorbital rims to include the anterior aspect of the maxilla and zygomatic buttress. The neurosurgeon then completes the craniotomy to remove the dysmorphic anterior cranial vault. With retraction of the frontal and temporal lobes of the brain (remaining anterior to each olfactory bulb), direct visualization of the anterior cranial base and orbits at the time of osteotomy is possible.
The orbital osteotomies are then completed across the orbital roof and superior aspect of the medial orbital wall, laterally through the lateral orbital wall, and inferiorly just into the inferior orbital fissure. The three-quarter orbital osteotomy units, with their tenon extensions, are removed from the field. The orbital units are reshaped and inset into a preferred position. Orbital depth is thereby increased, and ocular globe proptosis is reduced. Fixation is generally achieved with interosseous wires at each infraorbital rim and with microplates and screws at the tenon extensions and frontonasal regions.
The removed calvaria is cut into segments, which are placed individually to achieve a more normally configured anterior cranial vault. The goal of reshaping is to narrow the anterior cranial base and orbital width slightly and provide more forward projection and overall normal morphology.
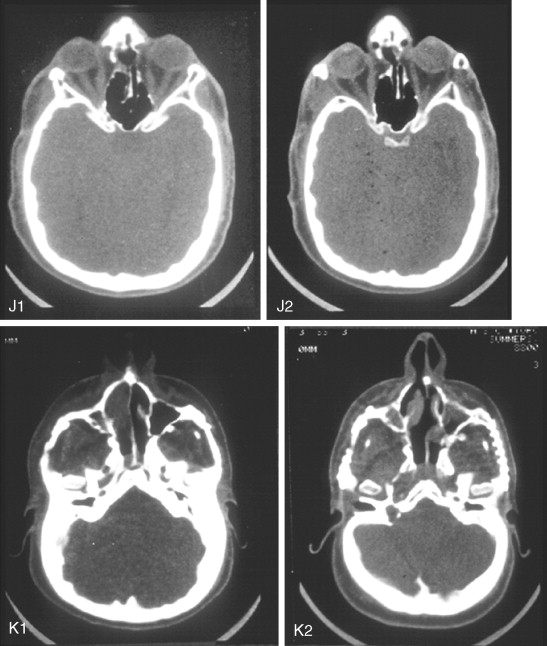
Further Craniotomy for Additional Brain Decompression: Cranial Vault Expansion and Reshaping in Young Children
After the initial suture release, brain decompression and reshaping of the cranio-orbital region is carried out during infancy, the child is observed clinically at intervals by the craniofacial surgeon, pediatric neurosurgeon, pediatric ophthalmologist, and developmental specialist and undergoes interval CT scanning. Should signs of increased ICP develop, urgent brain decompression with further cranial vault expansion and reshaping is performed. When increased ICP occurs, the suspected location of brain compression influences the region of the skull for which further expansion and reshaping is planned.
If the brain compression is judged to be anterior, further anterior cranial vault and upper orbital osteotomies for expansion with reshaping and advancement are carried out. The technique is similar to that described previously ( Figure 45-1 ). If the problem is posterior brain compression, expansion of the posterior cranial vault, with the patient in the prone position, is required ( Figure 45-2 ).
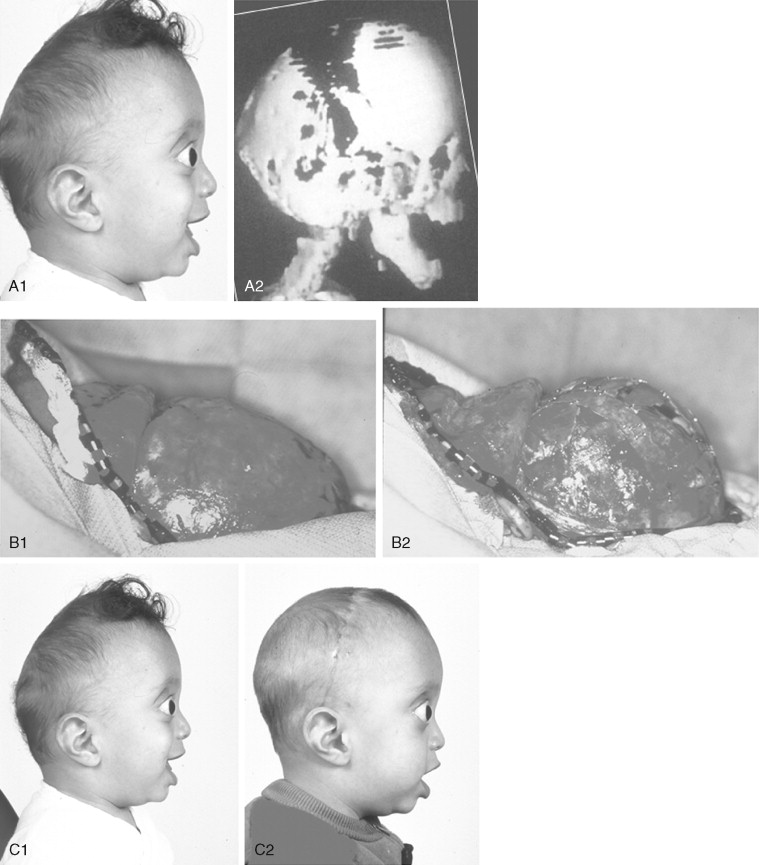
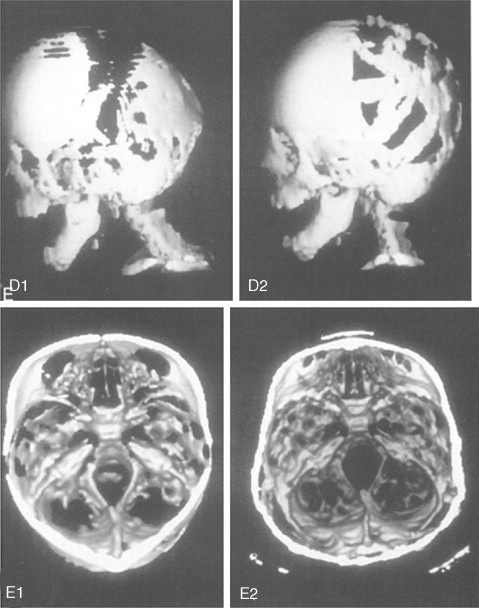
The “repeat” craniotomy carried out for further brain decompression and cranial reshaping in the child with Crouzon syndrome is often complicated by brittle, irregular cortical bone (which lacks a diploic space and may contain sharp spicules piercing the dura), the presence of previously placed fixation devices in the operative field, and convoluted thin dura compressed against (or herniated into) the inner table of the skull. All of these problems result in a greater potential for dural tears during the calvarectomy than would normally occur during the primary procedure. A greater potential for morbidity should be anticipated when reelevating the scalp flap, dissecting the dura free of the inner table of the skull and cranial base, and then removing the cranial vault bone.
Management of “Total Midface” Deformity in Childhood
The type of osteotomies selected to manage the “total midface” deficiency and/or deformity and residual cranial vault dysplasia should depend on the extent and location of the presenting dysmorphology rather than on a fixed universal (tunnel vision) approach to the midface malformation. *
* , , , , , , .
A main objective is to “normalize” the orbits, zygomas, and cranial vault, improving but not correcting the maxilla (at the Le Fort I level); correction of the maxillomandibular deformity will require orthognathic surgery. The selection of either a monobloc (with or without additional orbital segmentation), facial bipartition (with or without additional orbital segmentation), or Le Fort III osteotomy (intracranial versus extracranial) to manage the basic horizontal, transverse, and vertical upper face and midface deficiencies and/or deformities in a patient with Crouzon syndrome depends on the patient’s presenting midface and anterior cranial vault morphology. The observed dysmorphology is dependent on the original malformation, the previous procedures carried out, and the effects of ongoing growth.
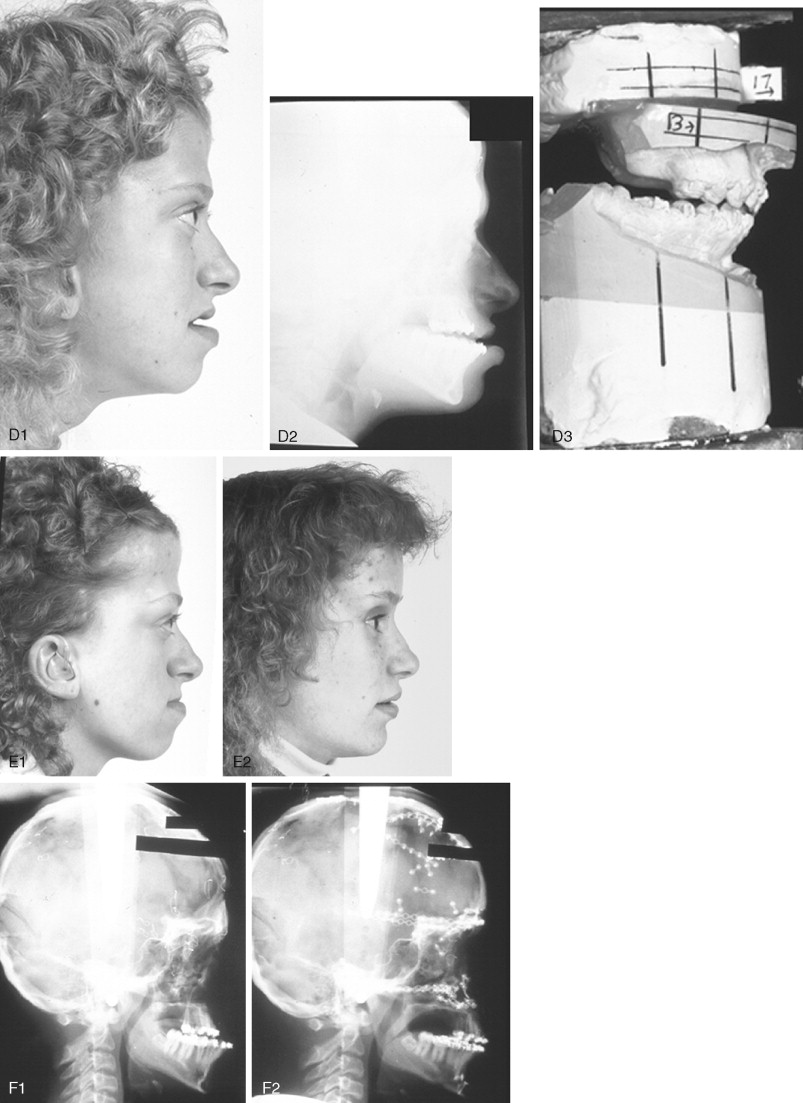
When evaluating the upper face and midface in a child born with Crouzon syndrome, if the supraorbital ridge is in good position when viewed from the sagittal plane (the depth of the upper orbits is adequate), the midface and forehead have a normal arc of rotation in the transverse plane (not concave), and the root of the nose is of normal width (minimal orbital hypertelorism), there is no need to further reconstruct this region (the forehead and upper orbits). In such patients, the basic residual midface deficiency and/or deformity is in the lower half of the orbits, zygomatic buttress, and maxilla. If so, the orbital-midface deformity may be effectively managed using an extracranial Le Fort III osteotomy ( Figure 45-3 ).
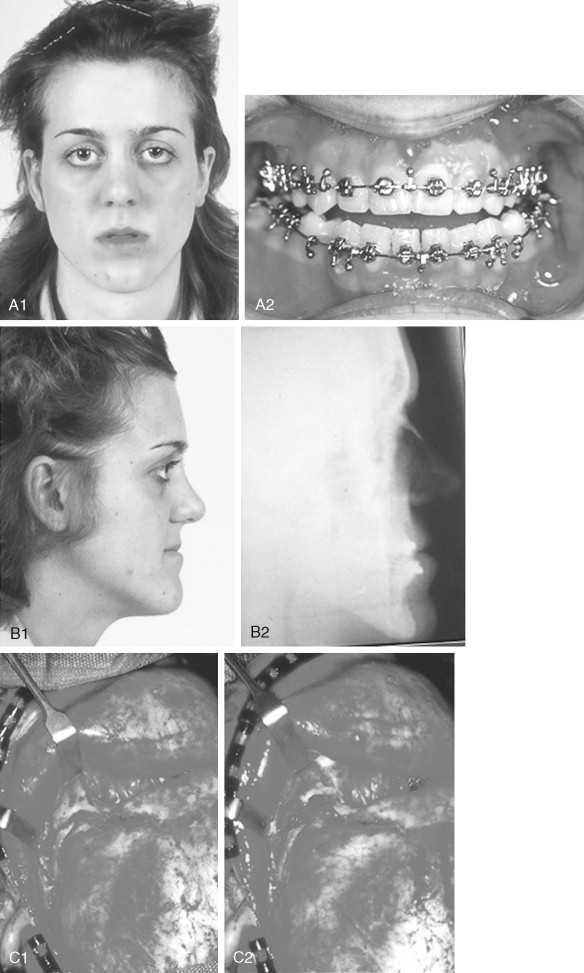
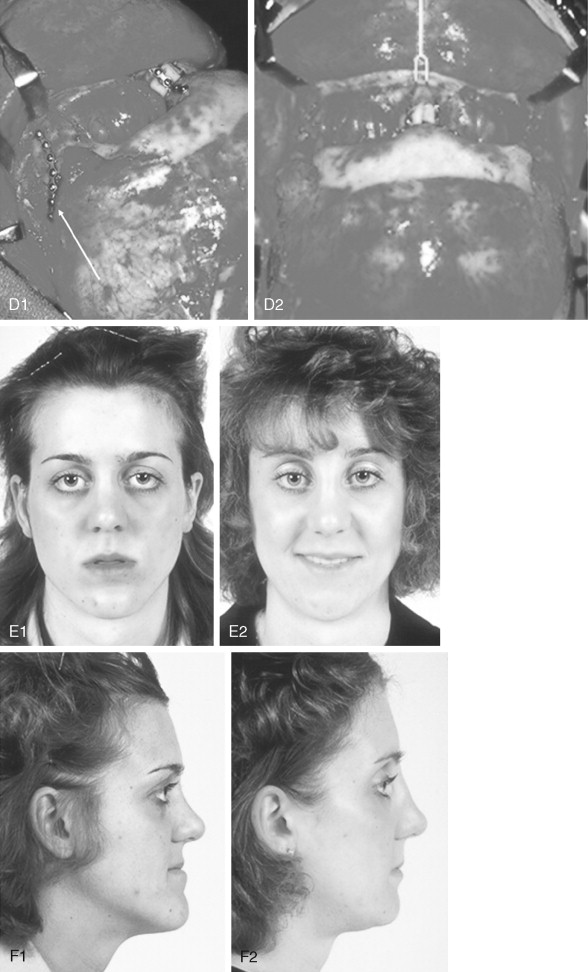
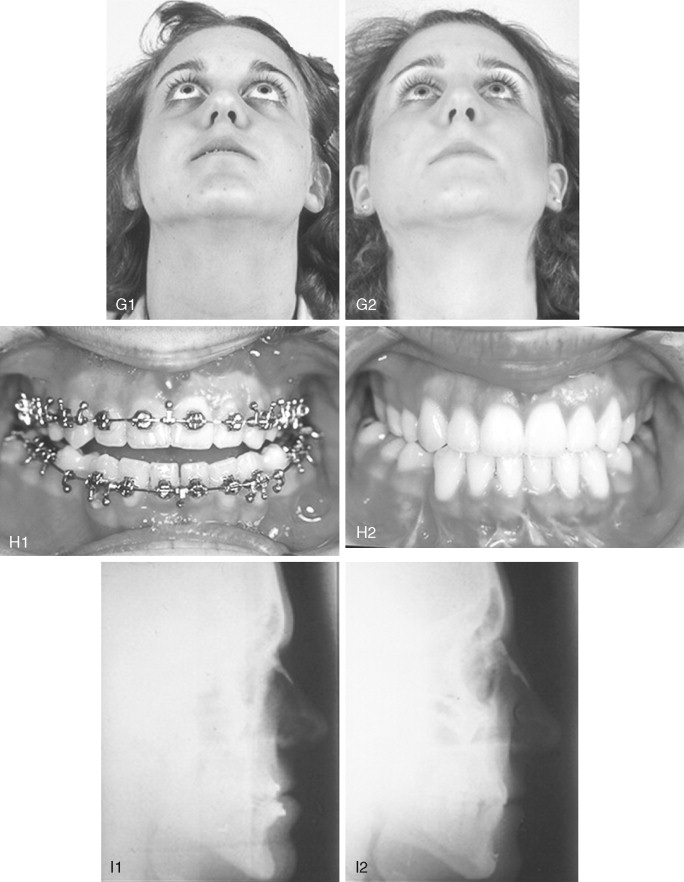
If the supraorbital region, anterior cranial base, zygomas, nose, lower orbits, and maxilla all remain deficient and/or deformed in the sagittal plane (horizontal retrusion), a monobloc osteotomy is indicated ( Figure 45-4 ). In these patients, the forehead is generally flat and retruded and will also require reshaping and advancement. If upper midface hypertelorism (increased transverse width) and midface flattening (horizontal retrusion) with abnormal facial curvature (concave arc) are also present, then the monobloc unit is split vertically in the midline (facial bipartition), a wedge of interorbital (nasal and ethmoid) bone is removed, the orbits and zygomas are repositioned medially while the maxillary posterior arch is widened. A facial bipartition is rarely required in Crouzon syndrome, but the monobloc is. When a monobloc or facial bipartition osteotomy is carried out as the “total midface” procedure, additional segmentation of the upper and lateral orbits for reconstruction may also be required to normalize the morphology of the orbital aesthetic units.
In most cases, an error in judgment will occur if the surgeon attempts to simultaneously adjust the orbits and idealize the occlusion using the Le Fort III, monobloc, or facial bipartition osteotomy in isolation, without completing a separate Le Fort I osteotomy. The degree of horizontal deficiency observed at the orbits and maxillary dentition are rarely uniform. This further segmentation of the midface complex at the Le Fort I level is required to establish normal proportions. If a Le Fort I separation of the “total midface” complex is not carried out and the surgeon attempts to achieve a positive overbite and overjet at the incisor teeth, an overadvancement of the orbits with enophthalmos will occur. The Le Fort I osteotomy is generally not performed at the time of the “total midface” procedure. This will await skeletal maturity and then be combined with orthodontic treatment. Until then an Angle Class III malocclusion will remain. Occasionally the teenager or adult will require both total midface and orthognathic management. The procedures may be carried out simultaneously ( Figure 45-5 ).
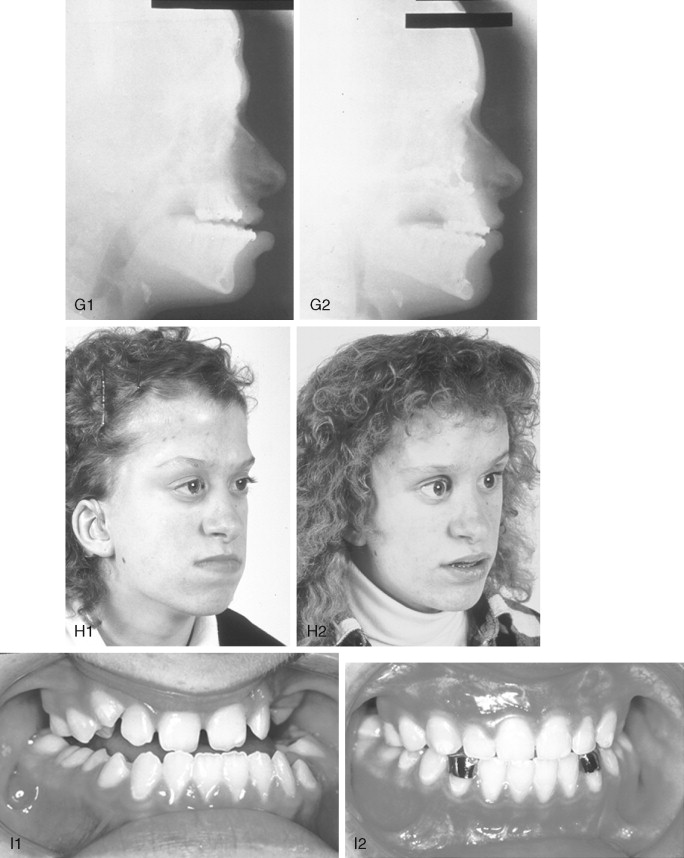
A major aesthetic problem specific to the Le Fort III osteotomy when its indications are less than ideal is the creation of irregular step-offs in the lateral orbital rims. This will occur when even a moderate (Le Fort III) advancement is carried out. These lateral orbital step-offs are unattractive and will be visible to the casual observer at a conversational distance; surgical modification (revision) performed later is difficult with less than ideal aesthetic results. Another problem with the Le Fort III osteotomy is the difficulty in judging an ideal orbital depth. A frequent result is either residual proptosis or enophthalmos. The simultaneous correction of orbital hypertelorism or correction of a midface arc-of-rotation problem is not possible with the Le Fort III procedure. Excessive lengthening of the nose, accompanied by flattening of the nasofrontal angle will also occur if the Le Fort III osteotomy is selected when the skeletal morphology favors a monobloc or facial bipartition procedure. It is not possible to later correct the surgically created vertical elongation and flattened nasofrontal angle of the nose.
Final reconstruction of the cranial vault deformities and orbital dystopia in Crouzon syndrome can be managed as early as 6 to 9 years of age. By this age, the cranial vault and orbits normally attain approximately 85% to 90% of their adult size. Whenever feasible, waiting until the maxillary first molars are erupted and in occlusion is preferred. When the “upper midface” and final cranial vault procedure is carried out after approximately 7 years of age, the reconstructive objectives are to approach adult dimensions in the cranio-orbito-zygomatic region, with the expectation of a stable result (no longer influenced by growth) once healing has occurred. Unfortunately, no further horizontal growth of the midfacial skeleton should be anticipated after a midface advancement procedure. Psychosocial considerations also support the age 6- to 9-year time frame for the upper midface and final cranial vault procedure. When the procedure is carried out at this age, the child may progress through school with a chance for a satisfactory body image and self-esteem. Routine orthognathic surgery will be necessary at the time of skeletal maturity to achieve an ideal occlusion and facial balance.
Orthognathic Procedures for Facial Balance and Definitive Occlusal Correction
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


