Introduction
Cherubism is a human genetic disorder that causes bilateral symmetrical enlargement of the maxilla and the mandible in children. It is caused by mutations in SH3BP2 . The exact pathogenesis of the disorder is an area of active research. Sh3bp2 knock-in mice were developed by introducing a Pro416Arg mutation (Pro418Arg in humans) in the mouse genome. The osteoclast phenotype of this mouse model was recently described.
Methods
We examined the bone phenotype of the cherubism mouse model, the role of Sh3bp2 during bone formation, osteoblast differentiation, and osteoblast function.
Results
We observed delays in early postnatal development of homozygous Sh3bp2 KI/KI mice, which exhibited increased growth plate thickness and significantly decreased trabecular bone thickness and bone mineral density. Histomorphometric and microcomputed tomography analyses showed bone loss in the cranial and appendicular skeletons. Sh3bp2 KI/KI mice also exhibited a significant decrease in osteoid formation that indicated a defect in osteoblast function. Calvarial osteoblast cell cultures had decreased alkaline phosphatase expression and mineralization, suggesting reduced differentiation potential. Gene expression of osteoblast differentiation markers such as collagen type I, alkaline phosphatase, and osteocalcin were decreased in osteoblast cultures from Sh3bp2 KI/KI mice.
Conclusions
These data suggest that Sh3bp2 regulates bone homeostasis through not only osteoclast-specific effects, but also through effects on osteoblast differentiation and function.
Cherubism is an autosomal dominant human genetic fibro-osseous disorder that affects children around 3 to 4 years of age. Symmetrical cyst-like cavities in maxillae and mandibles are the hallmark of cherubism. Cavities fill with soft fibrous tissue, which is histologically indistinguishable from giant cell granulomas. Expansion of the fibrous tissue can lead to characteristic facial deformities in cherubism patients. In most cases, the fibrous lesions regress spontaneously after puberty. Mild cases of cherubism can escape initial clinical diagnosis. Typically, patients exhibit multiple radiolucencies, more frequently in the mandible. Only in severe cases, after the active phase of the disorder, there is some facial deformity, and the patients might need surgical correction. Mutations for cherubism have been identified in SH3BP2 , which codes for an adaptor protein expressed in several cell types including osteoblasts and osteoclasts. Adaptor proteins usually lack enzymatic activity but are important for forming protein complexes by interacting with other proteins involved in signal transduction. The function of SH3BP2 is not well understood. Studies of T- and B-lymphoid cells suggest that SH3BP2 can act as a stimulator of nuclear factor of activated T-cells (NFAT)-mediated transcription. Other studies suggest that increased NFAT activity increases osteoclastogenesis and might regulate osteoblast proliferation.
To investigate mechanisms leading to cherubism, Ueki et al developed a knock-in mouse model by introducing a Pro416Arg mutation in SH3BP2 (Pro418Arg in humans) into the mouse genome. Homozygous knock-in ( Sh3bp2 KI/KI ) mice are osteopenic, with increased numbers of osteoclasts, and they exhibit increased macrophagic infiltration and significantly increased tumor necrosis factor-α (TNF-α). The results of Ueki et al suggest that bone loss in cherubism is in part due to the inflammatory effects of TNF-α, leading to increased osteoclastogenesis, which is consistent with a previous report showing that TNF-α can induce inflammatory bone loss via increased osteoclastogenesis. The role of the cherubism mutation on osteoblast differentiation and function was not addressed.
The goals of this study were to further characterize the bone phenotype of cherubism knock-in mice and to examine the role of Sh3bp2 in osteoblast differentiation and function for the first time.
Material and methods
Cherubism knock-in mice ( Sh3bp2 +/KI and Sh3bp2 KI/KI ) carry a Pro416Arg mutation in Sh3bp2 (equivalent to a Pro418Arg mutation in humans). For our experiments, Sh3bp2 +/KI mice were crossed with CD1 mice, and F2 litters (male mice) were used except for the quantitative polymerase chain reaction (QPCR) experiments, in which we used cells from C57B6 background. The Institutional Animal Care and Use Committee at the University of Connecticut Health Center, Farmington, approved all animal protocols.
We performed skeletal analysis after Alcian blue and alizarin red staining on postnatal animals at days 7, 14, and 21. Male and female mice were pooled for this experiment (n = 6). Briefly, the animals were defleshed, eviscerated, and fixed in 95% ethanol at room temperature for 7 days. The specimens were incubated in 0.015% Alcian blue in 95% glacial acetic acid overnight and subsequently treated in 95% ethanol for 2 days. Tissues were cleared in 1% aqueous solution of potassium hydroxide and incubated in 0.1% alizarin red in 1% potassium hydroxide overnight at room temperature. The samples were then passed through increasing concentrations of glycerol and stored in 100% glycerol at room temperature.
The skulls of 10-week-old male mice were defleshed and fixed in 4% paraformaldehyde (PFA) at 4°C (n = 6 for each group). Craniofacial morphology was assessed by using previously described guidelines. We measured skull length, skull width, inner canthal distance, and snout length and compared Sh3bp2 +/+ mice with Sh3bp2 +/KI and Sh3bp2 KI/KI mice. Measurements were obtained by using the measurement tool in Photoshop (version 7.0, Adobe, San Jose, Calif).
Dual energy x-ray absorptiometry (DEXA) was used on the skulls, mandibles, and femurs from 10-week-old male Sh3bp2 +/+ , Sh3bp2 +/KI , and Sh3bp2 KI/KI mice that were dissected and fixed in 4% PFA (n = 6 for each group). Bone mineral density (BMD) and bone mineral content (BMC) were measured by using a PIXImus densitometer (Lunar, Madison, Wis).
Microcomputed tomography (μ-CT) was performed on femurs, calvariae, and mandibles from 10-week-old male mice (n = 6) by using an mCT20 system (Scanco Medical AG, Bassersdorf, Switzerland). Individual bones were dissected, and adjacent soft tissue was carefully removed and fixed in 4% PFA. Trabecular measurements of femurs were taken at the distal growth plate in 80 consecutive slices of 12-μm resolution over a distance of 960 μm. Volumetric regions were rendered as 3-dimensional arrays with an isometric voxel dimension of 12 μm. Fifty cross-sectional slices of 12 μm in the middiaphysis were used to calculate cortical bone parameters. Mandibular cortical thickness was analyzed by measuring vertical sections at the mandibular foramen. Calvariae were analyzed over an area of 100 slices by using the sagittal suture of the central parietal region as a reference point.
We used femurs from the same mice as for the μ-CT (n = 6). Decalcified bones were embedded in paraffin. Longitudinal serial sections, 5 μm thick, were cut on a Polycut S microtome (Reichert-Jung, Heidelberg, Germany) with a D profile knife (Delaware Diamond Knives, Wilmington, Del). Sections were taken from the middle of the femur containing the central vein and were stained with modified Masson trichrome. Osteoblasts were identified as cuboidal cells lining trabecular bone. Osteoclasts were identified as multi-nucleated, tartrate-resistant acid phosphatase (TRAP) positive cells on the trabecular surface. Osteoblast and osteoclast numbers were normalized to trabecular bone surface.
Quantification of osteoid thickness and average growth plate thickness was performed on polymethylmethacrylate sections 5 μm thick by using Bioquant software (Biometrics, Nashville, Tenn). The measurements, terminology, and units were according to the recommendations of the nomenclature committee of the American Society for Bone and Mineral Research.
Calvariae from 2- to 7-day-old mice were dissected free of sutures, placed in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 IU per milliliter of penicillin, 100 μg per milliliter of streptomycin (all Gibco BRL, Grand Island, NY) and incubated overnight at 37°C in 5% carbon dioxide. Calvariae of the same genotype were subjected to 4 sequential 15-minute digests with an enzyme mixture of 1.5 U per milliliter of collagenase P (Roche Applied Science, Indianapolis, Ind) in phosphate-buffered saline solution and 0.05% trypsin per 1 mmol/L of EDTA (Gibco BRL) at 37°C on a rocking platform. The second to fourth digests were pooled, and the cell pellets were collected by centrifugation, resuspended in DMEM, and filtered through a 70-μm cell strainer (Becton Dickinson, Franklin Lakes, NJ). These fractions contained mainly osteoblasts and their progenitors. Cells were plated at a density of 1 × 10 4 cells per square centimeter in 24 well plates in DMEM containing 10% fetal bovine serum and antibiotics. Differentiation was induced by changing to α-MEM supplemented with 50 μg per millileter of ascorbic acid and 8 mmol/L of β-glycerophosphate (Sigma-Aldrich, St Louis, Mo) after a week. The media were changed every 2 days.
Calvarial osteoblasts were isolated and plated as described above. Cells in parallel plates were trypsinized and counted at days 1, 3, 5, and 7. For each group, cells were plated in triplicate, and 3 readings were taken for each well. The assay was repeated 3 times. For cell-death assays, cells were plated in chamber slides (Lab-Tek II, Thermo Fisher Scientific, Rochester, NY) at a density of 1 × 10 4 cells per square centimeter. When cells reached 50% to 60% confluency, they were deprived of serum for 3 days and incubated with 4 μg per milliliter of camptothecin for 3 to 4 hours to induce apoptosis. A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed according to the manufacturer’s protocol (Promega, Madison, Wis). DNase I treatment was used as a positive control, and no enzyme treatment was used as a negative control. The apoptosis rate was calculated as the ratio of TUNEL positive cells to the total number of cells by using the Photoshop software.
Mouse calvarial osteoblasts grown in differentiation medium for 14 and 21 days were subjected to alkaline phosphatase staining according to the manufacturer’s protocol (Sigma-Aldrich). Alkaline phosphatase-stained plates were further treated with von Kossa’s stain to detect mineralization. Briefly, samples were rinsed with deionized water, and a 5% silver nitrate (Sigma-Aldrich) solution was added and incubated under a light source for 40 minutes before quantification by using a flatbed scanner and the Photoshop software.
Primary osteoblast calvarial cultures were differentiated, and cells were harvested on days 0, 7, 14, and 21. Total RNA was isolated by using Trizol reagent (Life Technologies, Grand Island, NY). cDNAs were prepared from total RNA by using Sprint Power Script Oligo dT (BD Biosciences, Palo Alto, Calif). cDNA messages were amplified by using an ABI7500 thermal cycler (Applied Biosystems, Carlsbad, Calif) and ABI SYBR Green Power Mix (Applied Biosystems). Mouse 18S RNA was used as a reference. Real-time QPCR analysis primer sequences are described in Table I .
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Col1a | 5′-ACGTCCTGGTGAAGTTGGTC-3′ | 5′-CAGGGAAGCCTCTTTCTCCT-3′ |
| Tnap | 5′-GCTGATCATTCCCACGTTTT-3′ | 5′-CTGGGCCTGGTAGTTGTTGT-3 |
| Bsp | 5′-CAGAGGAGGCAAGCGTCACT-3′ | 5′-CTGTCTGGGTGCCAACACTG-3′ |
| Runx2 | 5′-ACCTAGTTTGTTCTCTGATCGCCT-3′ | 5′-GGGATCTGTAATCTGACTCTGTCCTT-3′ |
| Ocn | 5′-AAGCAGGAGGGCAATAAGGT-3′ | 5′-TTTGTAGGCGGTCTTCAAGC-3′ |
| 18S | 5′-TTGACGGAAGGGCACCACCAG-3′ | 5′-GCACCACCACCCACGGAATCG-3′ |
Primary calvarial osteoblasts were isolated from newborn mice as described above and grown to confluency. Cells either freshly isolated or cultured in differentiation medium for 2 weeks were trypsinized and labeled with antibodies against CD11b, CD45, and Ter199 (e-Biosciences, San Diego, Calif) for flow cytometry (FACS) analysis (FACS Calibur, BD Biosciences, San Jose, Calif).
Statistical analysis
Each experiment was repeated 3 times, and data points for each experimental group were expressed as the mean ± standard error of the mean. The unpaired t test was used to determine the statistical significance between groups. P ≤0.05 was considered significant.
Results
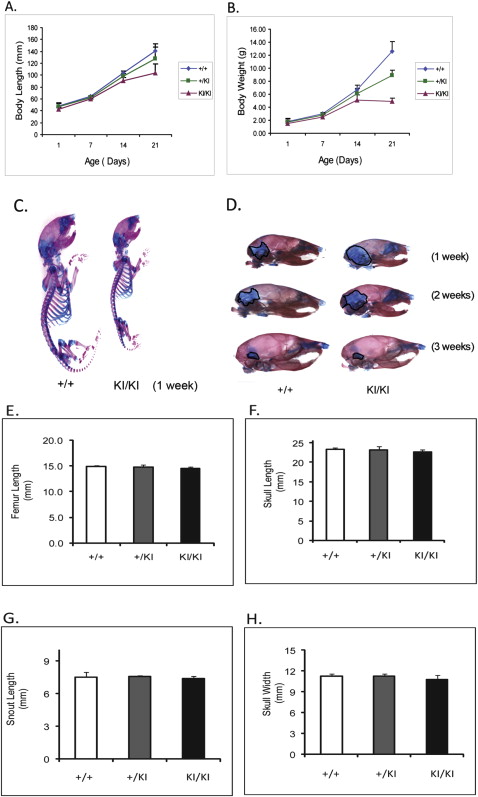
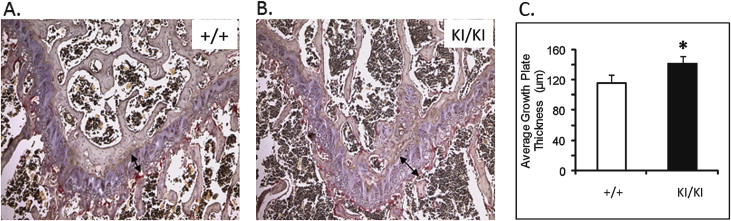
At 1 week of age, there was no detectable difference between the Sh3bp2 +/+ and Sh3bp2 +/KI mice in size and behavior, but the Sh3bp2 KI/KI mice appeared to be smaller ( Fig 1 , C ). At 3 weeks, we found a significant decrease in body length ( P ≤0.001) of the Sh3bp2 KI/KI mice ( Fig 1 , A ). They measured 26% less in body length, and the Sh3bp2 +/KI mice had a 9% reduction. There were reductions in body weight of 61% for the Sh3bp2 KI/KI mice and 29% for the Sh3bp2 +/KI mice ( Fig 1 , B ). Alcian blue (cartilage) and alizarin red (bone) staining showed a slight delay in ossification ( Fig 1 , D ). The Sh3bp2 KI/KI mice retained cartilage in the occipital and parietal regions of the skull for the first 2 weeks of postnatal development. This delay in ossification was not noticeable by 3 weeks of age. Morphologic evaluation of femur lengths and skull measurements showed no significant decreases in the Sh3bp2 +/KI and Sh3bp2 KI/KI mice ( Fig 1 , E-H ). On analyzing the growth plate of 10-week-old femurs, we found significant increases in growth plate thickness in the Sh3bp2 +/KI (8%) and Sh3bp2 KI/KI (21%) mice compared with the Sh3bp2 +/+ mice ( Fig 2 ).
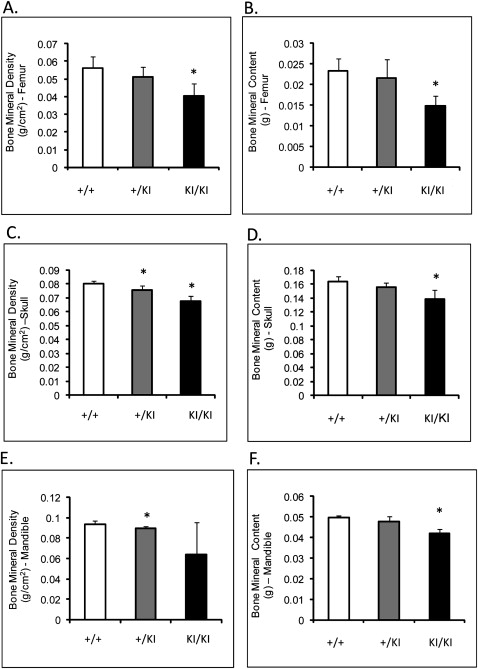
Femoral BMD (measured by DEXA) of the Sh3bp2 KI/KI mice was decreased by 27% ( P ≤0.001), and femoral BMC was decreased by over 36% ( P ≤0.0002) compared with the Sh3bp2 +/+ littermates ( Fig 3 , A and B ). The skull BMD and BMC of the Sh3bp2 KI/KI and Sh3bp2 +/KI mice were significantly decreased by about 15% compared with the Sh3bp2 +/+ skulls ( Fig 3 , C and D ). Mandibular BMD and BMC were reduced by 31% and 14%, respectively, in the Sh3bp2 KI/KI mice ( Fig 3 , E and F ). Overall, all bones examined in the mutant animals had decreased BMD and BMC.
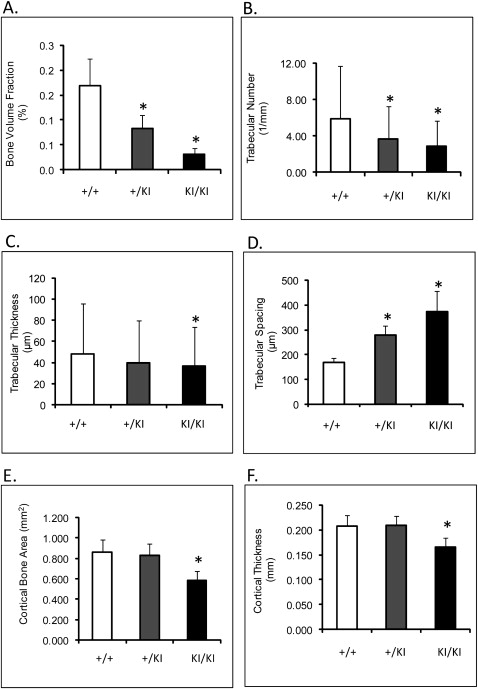
The μ-CT showed an 80% ( P ≤0.0013) decrease in femoral bone volume fraction of Sh3bp2 KI/KI mice compared with the Sh3bp2 +/+ mice ( Fig 4 , A ). Trabecular thickness was reduced by 31% ( P ≤0.014), and trabecular numbers decreased by 51% ( P ≤0.0001) ( Fig 4 , B-D ). Cortical thickness was reduced by 20%, and cortical bone surface area was reduced by 32% in the Sh3bp2 KI/KI animals ( P ≤0.01) ( Fig 4 , E and F ). Mandibles and calvariae also exhibited similar patterns of bone loss ( Tables II and III ). The 10-week-old Sh3bp2 KI/KI mice described here (C57B6/CD1 F2 mice) had severe bone loss. These results are consistent with previous results from mice homozygous for the knock-in mutation and crossed into C57B6 background.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


