Fig. 11.1
Cross section of blood vessels. Small blood vessels such as capillaries, postcapillary venules and arterioles are lined by a single layer of endothelial cells (ECs), surrounded by a continuous basement membrane (BM). Pericytes (perivascular cells) surround the basement membrane, and are themselves encircled by nearby stromal fibroblasts. The BM underlies the endothelial basal lamina histologically but is biochemically indistinguishable from the reticular fiber matrix synthesized by the pericytes. (Modified from Lafleur MA, Handsley MM and Edwards DR (2003) “Metalloproteinases and their inhibitors in angiogenesis,” Exp. Rev. Mol. Med. 5(23):1–39, Cambridge University Press: Copyright permission granted by Cambridge University Press, 2008)
The cellular elements of blood, the erythrocytes, leukocytes, and platelets are immersed in blood plasma, a rich solution of proteins, small molecules, and minerals. Most plasma proteins are made and secreted by the liver, but a few are made and secreted by the blood vessel endothelial cells. The cellular elements of blood are made from precursors in the red marrow of long bones (Sect. 9.2.1) by hematopoiesis. Aged cells are removed from the circulation by macrophages in the spleen, liver, or bone marrow. Platelets are derived from megakaryocytes, large cells that develop in the bone marrow along with erythrocytes and leukocytes. Platelets are outer membrane-covered secretion granules of megakaryocytes and are removed by macrophages passing through the spleen after only 8–12 days in circulation. Launching the blood coagulation system requires an interaction between plasma proteins and platelets at a site of injury.
11.1.1 Bleeding and Blood Clotting Problems
Diseases or drugs that affect the production of platelets or plasma proteins responsible for clotting cause excessive bleeding problems. The most common are diseases of the bone marrow and chemotherapy, both of which deplete platelets by inhibiting the production of megakaryocytes. In addition, liver disease, commonly caused by chronic alcoholism or viral infections, inhibits the production of the proteins responsible for blood coagulation. Less commonly, genetic mutations of these proteins cause life-long excessive bleeding (hemophilia). Finally, although chronic inflammation such as that associated with chronic periodontitis enhances clotting systemically, it often inhibits blood clotting locally (Sects. 13.2.3 and 13.3.1). Any of the above conditions may cause an unexpectedly large amount of bleeding from a tooth extraction and it is advisable to take a medical history and ensure that local inflammation is reduced as much as possible beforehand (Sect.13.5.1).
Capillaries are fragile. Continuous movement of the heart or intermittent movements and pressure changes around the cartilage between the bones stresses the capillaries, predisposing them to wear and tear that can activate the clotting system. Micro-size clots continually form and dissolve. A more severe injury such as bumping into a hard object causes bruising from bleeding beneath the skin. Although bleeding stops after a few minutes because a clot forms, the bruise lasts for a day or longer while leukocytes attracted by proinflammatory cytokines released by the damaged tissues (Sect. 13.3.2) digest the extravascular erythrocytes (Sect. 13.2.4). As the bruise disappears and the damaged tissue begins to be repaired, the clot is gradually lysed.
In older adults, there is a predisposition to clotting because of blood vessel degeneration or a decreased blood flow. The clots may obstruct a small artery or detach as an embolus that obstructs a arteries or veins elsewhere. Possible consequences are heart attack, stroke, pulmonary obstruction, or peripheral necrosis (Sect. 13.4.3). A tooth extraction in an elderly individual can cause an embolism elsewhere in the body. Elderly patients and patients on medication should be readied for oral surgery only after taking a history to elicit potential bleeding problems and consulting their attending physician. Rarely, acute disseminated intravascular blood clotting may be induced in any individual by a severe crushing injury, a severe allergic reaction to an insect bite or drug, or uncontrolled systemic bacterial or viral infections.
Summary
Capillaries are composed of endothelial cells and an outer pericyte layer that expresses contractile smooth muscle actin within a reticular fiber matrix consisting of thin collagen fibers surrounded by collagen and fibrillin microfibrils. The matrix is held together by proteo-glycosaminoglycans and anchored to the capillary basal lamina. Launching the blood coagulation system requires that platelets be present and activated at an injured site. Most blood clotting proteins are made in the liver; blood cells and platelets are made in the red bone marrow. Diseases of the liver or bone marrow thus enhance bleeding by removing clotting components. Blood vessel degeneration predisposes especially to heart attacks, strokes, pulmonary obstruction, and peripheral necrosis. Disseminated intravascular blood clotting is an often fatal condition induced by a severe crushing injury, allergy or infection.
11.1.2 Blood Vessel Injury, von Willebrand Factor, and Platelets
Platelets initiate coagulation by interacting with von Willebrand Factor (VWF), a multidomain protein (Fig. 11.2) synthesized and secreted by healthy endothelial cells. Following loss of signal peptide in the endothelial cell endoplasmic reticulum (ER), the proVWF molecule dimerizes by disulfide bonding at the C-terminal cysteine knot domain, which is homologous to the domain in TGF-β (Sect. 3.2.2). The dimers are transported to the Golgi, where propeptide domains D1 and D2 are cleaved, permitting large multimers to assemble by disulfide bond exchange between the dimer D3 domains. Sites of stress or injury expose subendothelial stromal collagen that binds to A3 domains in the multimer. The collagen interaction exposes A1 domains that tether platelets from the blood flow by binding to a platelet surface glycoprotein (Gp) called GpIbα. Tethering activates the platelets to aggregate into a platelet plug or soft clot which temporarily stops the bleeding and provides a surface for the more permanent blood clot. Platelet plug formation is enhanced mildly by epinephrine and ADP (Sect. 11.6.6), and strongly if thrombin is already activated (Sect. 11.3.4). Epinephrine is released into the blood as a response to injury, and ADP is released passively from injured endothelial cells and erythrocytes prior to activation. The activated platelets form a plug in three ways: First, an integrin on the platelet surface (αIIbβ3) is activated and binds to an RGD sequence (Sect. 3.2.1) on the VWF C1 domain (Fig. 11.2), but is displaced by fibrinogen (1.5 – 3.0 mg/ml in blood plasma, 100 – 300 times greater than multimeric VWF); Second, phosphatidylserine is moved from inner to outer leaflet of the platelet membrane; Third, granules whose contents stimulate thrombosis (blood clotting) and wound healing are secreted. One secreted component is thromboxane A 2 (TXA2), an eicosanoid (Fig. 13.13.) that stimulates thrombosis. TXA2 recruits and activates additional platelets from blood without VWF binding. Fibrinogen binds to the activated αIIbβ3 integrin, enlarging the soft clot (plug) at the injured site (Fig. 11.3). The activated platelets further seal capillaries by contracting actin in the pericyte cytosol, and the many platelets in the soft clot provide a large phosphatidylserine surface for coagulation. Multimeric VWF is therefore the sensor of blood vessel injury; it aggregates and activates platelets to plug the wound and launch the blood coagulation system.
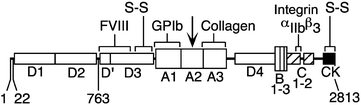
Fig. 11.2
Structure of von Willebrand factor (VWF). The VWF precursor (Expasy accession No. P04275) consists of a signal peptide (residues 1–22) and a propeptide (residues 23–763). The remaining residues 764–2,813 comprise the mature protein, which is divided into A, B, C, D and cysteine knot (CK) domains. The domains are named for sequence homology A1, A2 and A3 are homologous (similar) but unrelated to B, C and D etc. The CK domain is illustrated in detail in Fig. 12.4b. The domains were identified by related repeating sequences in this and many other proteins. Proteins with VWF domains relevant to topics in this book are type VI collagen (legend to Fig. 4.10) and salivary mucin MG1 (Sect. 12.3.1). The VWF binding sites for collagen (A3), platelet glycoprotein Ib, GPIb (A1), integrin αIIbβ3 (C1) and blood coagulation factor VIII are indicated. The down-pointing arrow indicates the site of degradation by ADAMTS-13 between tyr1605 and met1606 (From Sadler JE (2005) “New concepts in von Willebrand disease.” Annu. Rev. Med. 56:173–191. Reprinted, with permission, from the Annual Review of Medicine, Volume 56 ©2005 by Annual Reviews (www.annualreviews.org))
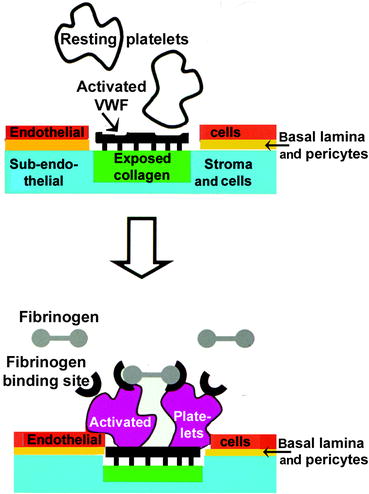
Fig. 11.3
Interactions of platelets with a wounded capillary surface. Top half: Resting platelets (white) possess glycoprotein-Ibα. This glycoprotein ligand recognizes a receptor on multimeric von Willebrand Factor (VWF) that becomes attached to exposed collagen (black) after injury has broken the endothelial cell layer and basal lamina (red). Bottom half: Upon contact, platelets are activated (maroon) and their surface undergoes two major changes during activation: (i) a fibrinogen receptor (black semicircle) appears and (ii) the phospholipids flip-flop (not shown). The fibrinogen receptor is activated integrin (αIIbβ3) which binds to a domain on each end of the linear fibrinogen molecule (details of the fibrinogen structure are given in Fig. 11.9). The lipids serve as a surface for localizing and activating clotting factors (Original figure submitted by Dr. Paul DeAngelis, Department of Biochemistry, University of Oklahoma HSC, Oklahoma City, OK, USA)
Multimeric VWF is degraded by an adamalysin, ADAMTS-13, which is secreted into the blood from endothelial cells along with VWF. ADAMTS-13 resembles procollagen N-peptidase (ADAMTS-2; Fig. 8.7b) except that the C-terminal domain of ADAMTS-2 is replaced by four additional thrombospondin-1 repeats (TS-1 repeats) and terminates in two CUB repeats (like those found on procollagen C-terminal peptidase, PCP, Fig. 8.7a). Mutations of ADAMTS-13 prevent the degradation of VWF. The concentration of multimeric VWF is increased, oversensitizing the blood to minor injuries at sites such as the heart where capillaries are stressed. The result is thrombotic thrombocytopenic purpura, a life-threatening disease in which small clots form, break loose, and block arterioles in the body. In contrast, mutations of VWF prevent it from binding collagen and do not stimulate platelets. The blood clotting system is underactivated and injuries cause excessive bleeding (hemophilia).
11.1.3 The Gamma-Carboxyglutamate Domain: A Calcium Ion Chelator
Activated platelets possess a phosphatidylserine surface that binds calcium ions (as in matrix vesicles; Sect. 9.3.3). This surface attaches various proteins that circulate in the blood and possess a negatively charged Ca+2 binding domain of up to nineγ-carboxyglutamate (gla) residues (Fig. 11.4). Osteocalcin (Sect. 9.4.2.) also possesses this gla domain.
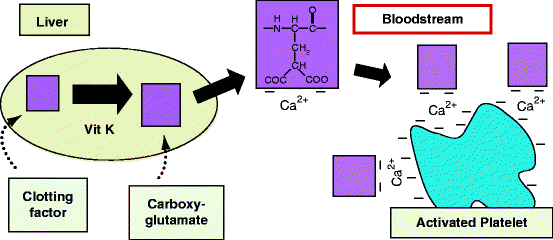
Fig. 11.4
Mechanism of clotting factor localization to an activated platelet surface. Left: After synthesis in the liver, certain blood clotting proteins are posttranslationally modified in the endoplasmic reticulum by a vitamin K-dependent Vit K carboxylase. This enzyme forms carboxyglutamate residues (top center) that chelate calcium ions. Right: In the bloodstream, clotting factor-bound calcium ions attach to negatively charged phosphatidylserine that appears on the surface of activated platelets. Certain therapeutic drugs or acquired deficiencies inhibit this process – see text (Original figure submitted by Dr Paul DeAngelis, Department of Biochemistry, University of Oklahoma HSC, Oklahoma City, OK, USA)
A gla protein is secreted into the endoplasmic reticulum (ER) of the liver where its N-terminal domain binds to and activates an enzyme that converts glutamic acid residues in the gla domain to γ-carboxyglutamic acid. The activated carboxylase enzyme then binds to a reduced form of vitamin K, a diphenol (KH2) to which it adds molecular oxygen. The product is an epoxide which reacts with a catalytic amine (N:) on the carboxylase (Fig. 11.5a), converting it to a strong epoxide base (K−) that abstracts a proton from the γ-carbon of a glutamyl residue in the gla domain. The resulting γ-carbanion spontaneously binds to carbon dioxide, converting it to a carboxyglutamate residue (Fig. 11.5b). Simultaneously, the proton (Fig. 5.11a) converts K− into a stable quinone epoxide, KO (Fig. 11.5b). Vitamin K oxidoreductase, a separate enzyme from the carboxylase in the ER membrane, completes the cycle by regenerating KH2 from KO. The carboxylase and oxidoreductase iteratively convert all the glutamate residues in a gla-domain. The carboxylase then catalyzes a proteolysis reaction that cleaves the N-terminal propeptide, releasing both carboxylated protein and propeptide separately. Carboxylation of glu to gla resembles oxidation of proline to hydroxyproline by vitamin C during collagen synthesis (Sect. 7.3.1.). The electrons may be obtained from glutathione and passed through an unknown reductase to a cystine disulfide bond in a small hydrophilic loop of vitamin K oxidoreductase protruding into the ER lumen. The electrons are relayed to a second cystine disulfide bond in the membrane of the oxidoreductase before being passed to KO.
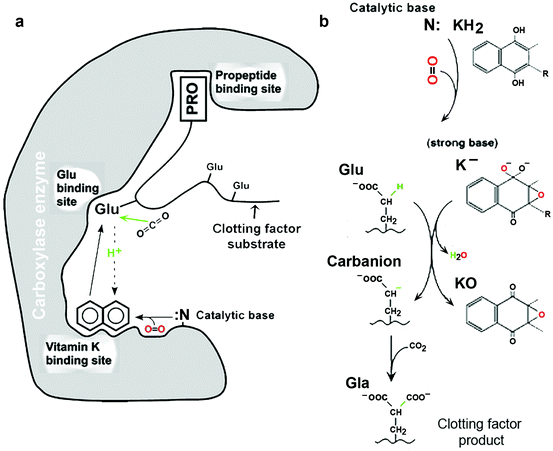
Fig. 11.5
Vitamin K-dependent carboxylase action. (a) Overall view of the carboxylase. The vitamin K-dependent protein substrates have high- and low-affinity domains for the carboxylase enzyme. The high affinity site is the propeptide, which is cleaved after carboxylation, and the low affinity site is the most N-terminal glutamate residue to be carboxylated. Carboxylation is initiated after the second site, the vitamin K binding site, is also occupied by reduced vitamin K (KH2). A catalytic amine (N:) deprotonates KH2, hydroquinone. A strong base epoxide (K−) forms and attacks a glu residue in the substrate binding domain (upward pointing solid arrow). Once this residue is carboxylated, the reactivity of N: is increased (downward pointing dotted arrow) and a tightly coordinated complex enables multiple glutamate residues in the substrate to be carboxylated by a processive mechanism (b): Vitamin K oxidation. When the strong base epoxide (K−) generates a glutamayl carbanion that reacts with CO2 to form gla, the base K− is oxidized to an intermediate that immediately loses water and forms a quinone epoxide (KO). KO regenerates KH2 by accepting electrons from glutathione via vitamin K oxidoreductase (Sect. 7.4.1). (From Figs. 1 and 2 in Berkner KL, (2005) “The Vitamin K-Dependent Carboxylase.” Annu. Rev. Nutr. 25:127–149. Reprinted, with permission, from the Annual Review of Nutrition, Volume 25 ©2005 by Annual Reviews (www.annualreviews.org))
Wafarin is one of a family of vitamin K mimics (coumarins) that successfully compete for the KH2 binding site on vitamin K oxidoreductase. The diphenol cannot be regenerated from KO, and glutamate is not carboxylated. The coumarins are therefore anticoagulants because the gla proteins are secreted into blood as precursors (Coumarins and problems associated with their use are discussed in Sect. 11.6.3) The clotting factor pro-proteases that possess a gla domain are factors II, VII, IX, and X (Table 11.1). These and other factors are carried around in the blood until they touch an activated platelet. The negative charge on the gla domain of the pro-protease completes a calcium sandwich with phosphatidylserine on the platelet aggregate. The negative lipid is on one side, the γ-carboxyglutamate is on the other, and the Ca2+ ions are in the middle (Fig. 11.4). Binding alters the conformation of the pro-protease factors and activates proteolysis.
Table 11.1
Major coagulation factors by pathway
|
Extrinsic
|
Intrinsic
|
Common
|
Inhibitory
|
|---|---|---|---|
|
Tissue factor (TF) (FIII)
|
FXIIb
|
VWF
|
Protein Ca
|
|
FVIIa
|
FXIb
|
Ca2+ ions (FVa)
|
Protein Sb
|
|
FIXa
|
FXa
|
||
|
FVIII
|
FV (FVa = FVI)
|
||
|
Prothrombin (FII)a
|
|||
|
Fibrinogen (FI)
|
|||
|
Transglutaminase (FXIII)
|
Summary
Collagen fibers exposed to blood after an injury are detected by Von Willebrand Factor (VWF), a plasma protein that circulates as multimeric aggregates. On binding collagen, VWF tethers platelets and activates an integrin that binds to an RGD site on the VWF aggregate, but more strongly to fibrinogen. VWF is degraded by an adamalysin, mutations of which enhance clotting. Mutations of VWF enhance bleeding. VWF adherent platelets release thromboxane, activating by-passing platelets to attach fibrinogen by their integrin receptors and a platelet plug or soft clot develops. The platelets in the soft clot expose a phosphatidylserine surface onto which calcium ions attach blood clotting proteins possessing γ-carboxyglutamate (gla) residues. Processing to gla occurs in the liver where a carboxylase binds to the N-terminal domain of these proteins and converts an internal domain rich in glutamate residues to gla residues. The carboxylase uses a derivative of vitamin K as a cofactor and oxidizes it to a quinone epoxide. The epoxide is reduced by a separate enzyme, vitamin K oxidoreductase, in tandem with gla domain carboxylation. After all of the domain’s glutamate residues are carboxylated, the carboxylase cleaves the substrate’s N-terminal propeptide, releasing the carboxylated protein and the propeptide separately. The oxidoreductase is inhibited by vitamin K analogues (coumarins) which cause the precursor proteins to be released without carboxylation and unable to bind to calcium ions on a phosphatidylserine surface. Large concentrations of vitamin K reverse the coumarin inhibition by providing large amounts of the initial derivative and allowing the oxidized quinone epoxide to be excreted instead of reduced.
11.1.4 The Extrinsic, Intrinsic, and Common Coagulation Pathways
Following the recruitment and activation of platelets by injured capillaries, their soft clot aggregates provide a surface for plasma proteins to interact and activate thrombin, which transforms soluble fibrinogen to a fibrin clot. Thrombin is activated by two blood coagulation pathways, extrinsic and intrinsic. A defective extrinsic path is incompatible with life, whereas defects of the intrinsic path cause hemophilia.
11.1.5 The Extrinsic Pathway
The extrinsic pathway is mediated by tissue factor (TF), also known as thromboplastin or factor III (Fig. 11.6a). Tissue factor is not present in blood plasma, i.e., it is extrinsic or outside of the vascular system. TF is a membrane protein that becomes exposed to the blood by trauma to the endothelial surface of blood vessels. It is especially rich in the membranes of pericytes, platelets, and leukocytes, but present in lesser amounts in most cells. It is absent from striated muscle cells and chondroblasts.
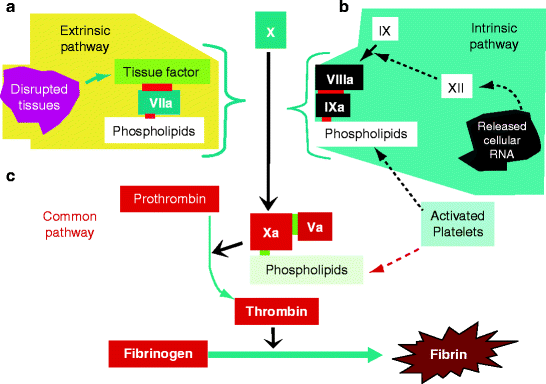
Fig. 11.6
Extrinsic and intrinsic pathways. (a) Extrinsic pathway (top left): When an injury activates platelets to form a phospholipid surface (white rectangle, lower right), disrupted tissues (magenta, top left) release tissue factor (TF) (green). Factor VII and its activated derivative (factor VIIa, blue) in blood plasma bind to the phospholipid surface by their gla domain and attach tissue factor. Unconverted factor VII is activated by small amounts of VIIa present and bound to TF. Factor X (dark blue) binds similarly to phospholipid and is converted by the TF–VIIa complex to Xa (red). (b) Intrinsic pathway (top right): Injury releases RNA, a negatively charged polymer that converts factor XII to XIIa (white squares). Factor XIIa activates factor XI (not shown), which remains soluble and activates factor IX bound to phospholipids (black square). Factor IXa activates bound factor X so that it weakly converts prothrombin to thrombin. The thrombin then activates factor VIII to VIIIa (black rectangle), which binds to factor IXa and accelerates its conversion of phospholipid-bound factor X. (c) Common Pathway (bottom center): Increasing amounts of thrombin activate factor V, converting it to Va. Va (small red square) binds to factor Xa (large red square) on the phospholipid surface and greatly accelerates the conversion of prothrombin to thrombin (red rectangles), and of fibrinogen (red rectangle) to fibrin (brown star). Factor numbering describes discovery, not interaction. More abundant factors were discovered and numbered first, fibrinogen (factor I), thrombin and prothrombin (factors IIa and II). Today, these factors are usually referred to by their names for clarity. The later-discovered factors are still referred to by their numbers. Factor XII was the last factor to be discovered because it is first in the cascade and present in least amounts in blood. A few molecules of factor XII activate more molecules of factor XI and factor IX, causing the intrinsic path to activate factor Xa rapidly (a, b, and c are original figures submitted by Dr. Paul DeAngelis, Department of Biochemistry, University of Oklahoma HSC, Oklahoma City, OK, USA)
When TF is exposed to blood, it spontaneously binds factor VII, a serine protease of the coagulation system that possesses a gla domain (Table 11.1). The TF–VII complex therefore adheres to the negatively charged surface of activated platelets. About 0.4% of factor VII is activated in blood by fatty acid- or triglyceride-mediated activation of factor IX and binds similarly to TF. The TF–VIIa complex (Fig. 11.6a
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


