Fig. 7.1
Collagen synthesis and processing in the endoplasmic reticulum. (a) Synthesis and processing of procollagen. Steps 1 through 4 are explained in the text. The N-terminal propeptide (green on left) contains a short triple helical region that is removed with the propeptide. The C-terminal propeptide (green) is larger than the N-terminal propeptide. The tropocollagen region (red) is much larger than illustrated. Blue hexagons indicate position of the hydroxylated lysine residues with attached glycan. (b) Procollagen triple helix formation. Proline hydroxylase (PH, dark green oval) is part of the inner wall of the endoplasmic reticulum. It binds to individual polypeptides and catalyzes proline hydroxylation. Following hydroxylation, the β subunit of PH, disulfide isomerase (PDI), catalyzes the formation of trople helical tropocollagen domain by disugide bonding between cysteine residues on the C-terminal propeptides. Hsp47 (red oval) is a chaperone that binds to the disulfide bonded tropocollagen and completes the triple helix formation. Procollagen α-polypeptides fail to form a triple helix and aggregate in the endoplasmic reticular lumen if Hsp47 is absent. Once the triple helix has formed, the procollagen is moved into a transport vesicle and transferred to the Golgi. The N- and C-terminal propeptide regions are indicated as green circles and green ovals. (a) Revised and partially updated from Fig.19-47 in The Molecular Biology of the Cell, Alberts et al., 4th Ed. 2002. Garland Science, Taylor & Francis Group, New York. (b) Slightly modified from Fig. 6 in Ishida, Y., et al., Molecular Biology of the Cell, Vol. 17, 2347–2355, May 2006; Reprinted with permission from the American Society for Cell Biology; PDF access http://www.molbiolcell.org/cgi/reprint/17/5/2346)
The procollagen trimers are taken into transport vesicles which pass them to cis-Golgi cisternae (Fig. 7.2). There, they: (a) aggregate into dense material (procollagen bundles seen in electron micrographs); (b) lose their N-terminal propeptide, which re-enters the cytosol and inhibits the translation of additional collagen; and (c) are is sorted into secretory vesicles in the trans-Golgi. Secretion strongly activates propeptide removal extracellularly (Sect. 8.2.1), generating spontaneous tropocollagen fiber formation and cross-linking (Sect. 4.2.2.). The synthesis and processing of non-fibrillar collagens are similar, except that their N- and C-terminal propeptides remain attached for use in the polymeric assemblages (Sect. 5.1.2).
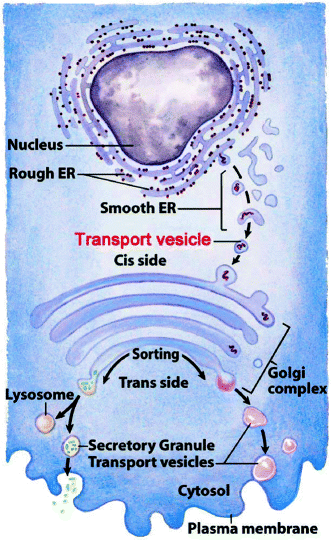
Fig. 7.2
Intracellular organelle path taken by collagen polypeptides before secretion. Diagram shows the rough and smooth endoplasmic reticulum, the budding of primary transport vesicles from the smooth endoplasmic reticulum to the cis-Golgi, the Golgi cisternae, and the budding of secretory or secondary transport granules (vesicles) from the trans-Golgi for secretion, delivery to lysosomes, or insertion into cell membranes from the trans-Golgi to form secretory granules or lysosomes (Slightly adapted from Fig. 27-35 in Lehninger Principles of Biochemistry. D.L. Nelson & M.M. Cox, 4th Ed. 2005. W.H. Freeman & Co., New York)
Summary
Collagen is synthesized in the endoplasmic reticular lumen. Each α-polypeptide possesses a signal sequence that guides the N-terminus into the ER where it is removed, leaving a long extended tropocollagen domain with small, folded N- and C-terminal propeptide extensions (procollagen). Proline hydroyxlase has two α subunits and two β subunits. The latter is called protein disulfide isomerase and it disulfide bonds C-terminal propeptides that have self-associated into trimers of two α1-chains and one α2-chain. Lysine is hydroxylated by an enzyme loosely homologous to the proline hydroxylase α-subunits. After selected proline and lysine residues are hydroxylated and hydroxylysine glycosylated, chaperone hsp47 replaces proline hydroxylase and guides triple helix formation of the tropcollagen domain. Procollagen then passes through the Golgi to secretory vesicles where it forms large procollagen bundles. The N-terminal propeptides are hydrolyzed and re-enter the cytosol where they inhibit collagen α-chain synthesis. Synthesis and processing of non-fibrillar collagens is similar, except that their N- and C-terminal propeptides are not removed.
7.1.1 Effects of Collagen Polypeptide Mutations
The fibrous collagens of all vertebrates and invertebrates possess about 340 gly-X-Y sequences encoded as groups of approximately 18 amino acids in their tropocollagen domain. The 41 to 42 exons are all multiples of 9 bases. Most are 54 bases in size, but some are also multiples of 54 bases, or combinations of 45- and 54-bases. The sequences of three such exons (Fig. 7.3), which suggest that the α-procollagen polypeptide evolved from a precursor of the N- and C-terminal domains by duplication of a 54 base exon that originally encoded 18 amino acids in the central portion of an ancient protein precursor of the procollagen N- and C-terminal domains. At about the same time, a small insertion within the C-terminal region enabled different α-polypeptide mixtures to associate. The resultant “molecular incestuous” mixing resulted in allowed the formation of collagen heterotrimers compatible with bone formation and the appearance of vertebrates from invertebrates. Invertebrate collagens are homotrimers and cannot calcify.

Fig. 7.3
Translated sequence of a tropocollagen exon. The18 amino acid residues (3×6 gly-X-Y sequence) encoded as a 54 bp exon. Sequence homologies between amino acids 1–18 in exon 1 of the tropocollagen domain of the COL1A1 gene compared with the sequence of 18 amino acid residues in exons 2 and 3 respectively. Numbering is according to den Dunnen JT and Antonarakis SE (2000). Human mutation, 15:7–12. Underlines show identity between exon 1 and exon 2 sequences. Highlights show identity between exon 1 and exon 3 sequences. The collagen genes are listed in Table 3.1. The COL1A1 gene encodes the type I α1 procollagen polypeptide and the COL1A2 gene encodes the type I α2 procollagen polypeptide.
Mutations that interfere with collagen fiber formation mostly cause lethal or nonlethal osteogenesis imperfecta, also known as brittle bone disease. The bones break easily and apparently spontaneously. The disorder occurs in about one in 50,000 live births in the US. Osteogenesis imperfecta is clinically divided by whether the teeth are also affected. They may appear opalescent blue-gray or yellow-brown because of abnormal dentin calcification. Occasionally, some teeth may be missing. Table 7.1 is a list of stromal protein diseases that affect tooth development.
Table 7.1
Stromal protein mutations affecting teeth
|
Mutationa
|
Disease
|
Symptom
|
|---|---|---|
|
Type I collagen mutations associated with osteogenesis imperfecta
|
Dentinogenesis imperfecta type I (see also Chap. 9)
|
Opalescent blue-gray or yellow-brown teeth because of abnormal dentin calcification
|
|
Kindlin-1 (Chap. 5)
|
Kindler syndrome
|
|
|
Unknown genes on chromosome 12
|
Ehlers–Danlos syndrome – type VIII
|
Aggressive periodontal disease
|
|
Laminin-5 (Chap. 5)
|
Junctional epidermolysis bullosa
|
Generalized enamel hypoplasia; increased caries
|
|
Fibrillin-1 and -2 (Chap. 6)
|
Marfan syndrome
|
Crowded incompletely developed (hypoplastic) teeth and deformities of the roots
|
|
Dentin sialophosphoprotein (DSPP), a glycoprotein of the connective tissue stroma (Chap. 9)
|
Dentinogenesis imperfecta type II
|
Changes resemble type I dentinogenesis imperfecta (Chap. 9)
|
|
Mutations of amelogenin formation and processing (Chap. 9)
|
Amelogenesis imperfecta
|
Brittle or absent enamel
|
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


