Molecular Biology of Host-Microbe Interactions
For the past two decades, the host response to the bacterial challenge that originates from the dental biofilm has been considered to play a major role in the initiation and tissue destruction of periodontal diseases.196 The significance of host-microbial interactions is reinforced by epidemiological data indicating different susceptibilities to periodontal disease among individuals, despite the long-term presence of oral biofilm.21,22,219 Other studies demonstrating increased susceptibility and greater severity of periodontal disease in individuals with impaired immune response due to systemic conditions also indicate the significance of the host response to the bacterial challenge.95,237 In the current paradigm of periodontal disease, specific periodontal pathogens are essential for disease initiation; however, the extent and severity of tissue destruction are largely dependent on the nature of the host-microbial interactions. These interactions are dynamic, because the microbial composition of the dental biofilm and host immune competency can vary widely among patients, thereby resulting in differences in host responses and subsequent alveolar bone loss. This concept has evolved in parallel with a more advanced appreciation of the immune response that results in an increased emphasis on mechanisms of host-microbial interactions in periodontal disease pathobiology as well as on the development of novel therapeutic strategies.
Periodontal diseases provide a unique situation for the study of microbial-host interactions. More than 500 different microbial species can be found in the oral biofilm268; however, only a few of those are associated with periodontal disease.332,333 This suggests that the recognition of both nonpathogenic/commensal bacteria and pathogenic bacteria by the host requires vigilance and tolerance mechanisms to mount an appropriate response that can prevent the dissemination of infection without inducing an exacerbated reaction that could result in damage to the host tissues. The direct recognition of microbes by the host is mediated by the recognition of microbial-associated molecular patterns (MAMPs) by pattern-recognition receptors (PRRs).35,36
The biological mediators expressed as a result of PRR-signaling activation include costimulatory molecules involved in the induction of adaptive immunity.36 This results in a cascade of events that will establish very complex cytokine and signaling networks. More recently, there is accumulating evidence for a direct role of “classical” innate immunity signaling through PRRs in adaptive immune cells that can modulate the function of these cells. Abundant evidence indicates a key role for the adaptive immune response—both the humoral and cellular aspects—in mediating the host response to microorganisms that exist within the oral biofilm as well as in the majority of tissue destruction associated with periodontal diseases.23,26,92,93,139,140 Although cells participating in the adaptive immune response are considered to be primary sources of cytokines, thereby leading to bone resorption,189 additional data exist that show how periodontal bone loss occurs in the absence of B and T cells, thus suggesting a role for the innate immune response in periodontal disease initiation or progression.24–26
Innate immunity and inflammation are not synonymous; however, inflammation arises primarily in response to infection. More importantly, innate and adaptive immune responses are not mutually exclusive, and the bridge between these arbitrarily and didactically distinguished arms of the immune response has been shortened in recent years. However, considering that this chapter focuses on host-microbe interaction and to avoid redundancy with other chapters, we will emphasize the direct recognition of MAMPs by cells participating in the immune response and the molecular mechanisms activated downstream from this recognition (Figure 9-1).
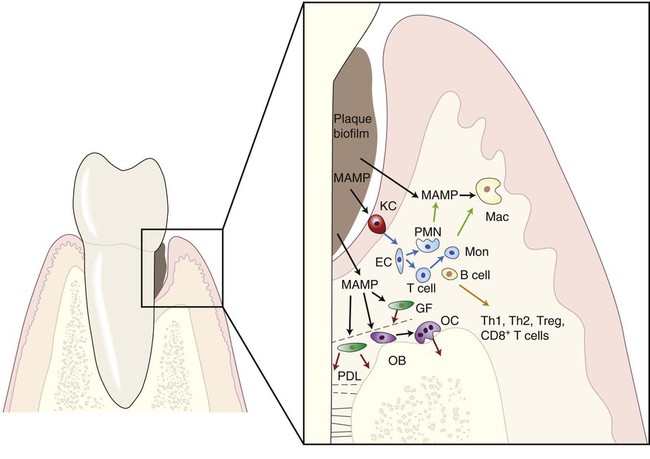
Innate Immunity in Periodontal Diseases
Vigilance and Tolerance
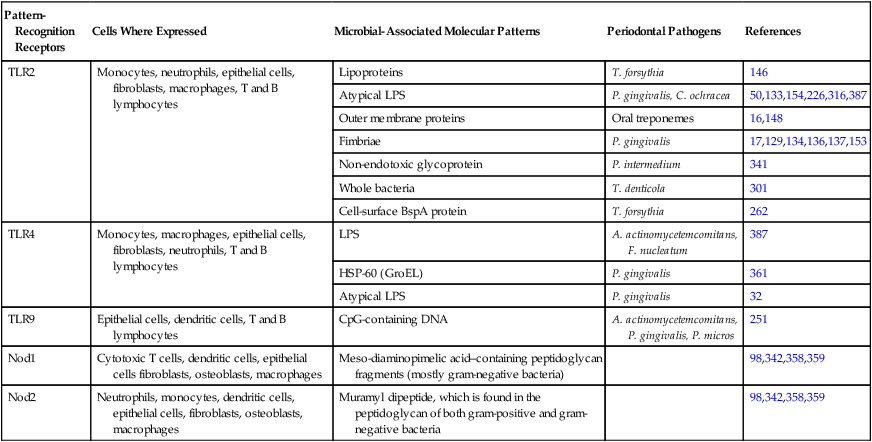
With regard to periodontal diseases, the current paradigm indicates that some groups or complexes of bacteria are more strongly associated with the presence of periodontal diseases. However, this association is not universally true, because there are individuals who harbor the disease-associated complexes that are free of periodontal disease. Moreover, the microbiota of the oral cavity can include more than 500 different species, and there is no uniformity with regard to the level of infection that is sufficient to produce disease. The complexity of the oral microbiota is particularly intriguing given the limited number of receptors that are able to recognize microbial antigens. Tolerance mechanisms probably play a role in modulating the host response to commensal/nonpathogenic bacteria. One of the primary challenges of the innate immune system is to discriminate among a large number of periodontal pathogens from the host with a limited number of cell surface receptors. This challenge is compounded because microbial pathogens have the ability to mutate as a mechanism of escaping host recognition. The innate immune system has met this challenge through the recognition of evolutionary conserved structures on pathogens that are not present in higher eukaryotes: the PRRs. These molecular motifs—the MAMPs—have essential roles in the pathogen’s ability to evade the host defense and thus are not subject to high mutation rates. MAMPs are shared among various species of microbes, but they are not expressed by the host. Although many PRRs have been known for years, it was not clear how the innate immune system functioned until the discovery of the Toll-like receptors (TLRs), which have proved to be critical for the recognition of microbes by the innate immune system and for bridging the innate and acquired immune responses. Interestingly, within the periodontal tissues, the expression of various TLRs appears to be increased in severe disease states.29 Table 9-1 illustrates PRRs and the host cells that express them and their ligands (i.e., the MAMPs) present in microorganisms.
TABLE 9-1
Pattern-Recognition Receptors and the Host Cells (Mouse or Human) That Express Them and Their Ligands (Microbial-Associated Molecular Patterns) Present in Microorganisms Relevant to Periodontal Disease
| Pattern-Recognition Receptors | Cells Where Expressed | Microbial-Associated Molecular Patterns | Periodontal Pathogens | References |
| TLR2 | Monocytes, neutrophils, epithelial cells, fibroblasts, macrophages, T and B lymphocytes | Lipoproteins | T. forsythia | 146 |
| Atypical LPS | P. gingivalis, C. ochracea | 50,133,154,226,316,387 | ||
| Outer membrane proteins | Oral treponemes | 16,148 | ||
| Fimbriae | P. gingivalis | 17,129,134,136,137,153 | ||
| Non-endotoxic glycoprotein | P. intermedium | 341 | ||
| Whole bacteria | T. denticola | 301 | ||
| Cell-surface BspA protein | T. forsythia | 262 | ||
| TLR4 | Monocytes, macrophages, epithelial cells, fibroblasts, neutrophils, T and B lymphocytes | LPS | A. actinomycetemcomitans, F. nucleatum | 387 |
| HSP-60 (GroEL) | P. gingivalis | 361 | ||
| Atypical LPS | P. gingivalis | 32 | ||
| TLR9 | Epithelial cells, dendritic cells, T and B lymphocytes | CpG-containing DNA | A. actinomycetemcomitans, P. gingivalis, P. micros | 251 |
| Nod1 | Cytotoxic T cells, dendritic cells, epithelial cells fibroblasts, osteoblasts, macrophages | Meso-diaminopimelic acid–containing peptidoglycan fragments (mostly gram-negative bacteria) | 98,342,358,359 | |
| Nod2 | Neutrophils, monocytes, dendritic cells, epithelial cells, fibroblasts, osteoblasts, macrophages | Muramyl dipeptide, which is found in the peptidoglycan of both gram-positive and gram-negative bacteria | 98,342,358,359 |
However, there are certain PRRs that can be secreted into the plasma as humoral proteins and others that are localized in the cytoplasm as intracellular sensors. Soluble PRRs include various proteins, such as collectins, ficolins and acute-phase pentraxins (e.g., C-reactive protein), which represent the functional ancestors of antibodies. These soluble mannose-binding receptors can interact with structures from microbes and activate the complement system via the mannan-binding lectin-associated serine kinase pathway.303 In fact, direct interaction between complement precursors and microbes can initiate activation through an unclear mechanism that will culminate in the opsonization and lysis of the microbes. Another example of soluble PRRs is the lectin-complement system, which is involved in recognizing the microbe by the carbohydrate-binding lectin that leads to its subsequent opsonization and phagocytosis.373
Nucleotide-oligomerization domain (NOD) protein-like receptors represent cytoplasmic PRRs, and they are characterized by C-terminal leucine-rich repeats (similar to the TLRs), an N-terminal caspase-activating recruitment domain, and a nucleotide-binding domain (i.e., a NOD). These were initially described as cytosolic TLRs and were analogous to the R proteins present in plants.76 NOD proteins are capable of recognizing different peptidoglycan molecules: Nod1 recognizes peptidoglycan that contains the meso-diaminopimelic acid fragments present in most gram-negative and some gram-positive bacteria,114 whereas Nod2 recognizes muramyl dipeptide, which is found in peptidoglycan from both gram-negative and gram-positive bacteria.235 More recent evidence indicates that NOD proteins (specifically Nod1 and Nod2) are involved in the activation of inflammatory gene expression98 and even in lipopolysaccharide (LPS) recognition independently of TLR.169 The relevance of NOD proteins for the immune response is demonstrated by the association between genetic mutations on Nod1 and Nod2 and the development of allergic conditions and Crohn’s disease, respectively.165,170 Another type of intracellular PRR is represented by another cytoplasmic protein family, the retinoic acid-inducible gene I–like receptors, which play a critical role in recognizing viral RNA as well as the induction of type I interferon expression. However, because the role of retroviruses in periodontal disease is not clear, these receptors will not be commented on here (for a review of retinoic acid-inducible gene I–like receptors, see Reference 385).
• Macrophages and polymorphonuclear cells, as professional phagocytes with the primary function of engulfing and destroying microbes;
• Dendritic cells, as professional antigen-presenting cells and activators of adaptive immunity; and
• Natural killer cells, as innate cytotoxic lymphocytes that recognize and kill host cells that are altered (e.g., tumor cells) or infected with viruses.
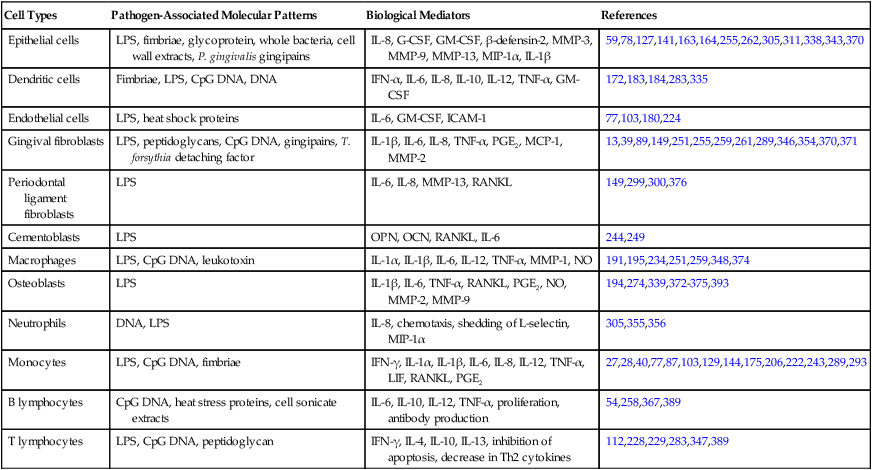
However, other cell types can also play important roles in innate immunity, because they are able to recognize MAMPs through their PRRs and to respond by expressing biologically active molecules (e.g., cytokines, matrix metalloproteases [MMPs]) that will have an effect on the homeostasis of the host tissues in the periodontal microenvironment. Resident “nonprofessional” cells (e.g., fibroblasts, osteoblasts) are also capable of producing a variety of cytokines (e.g., interleukin-6, prostaglandin E2, MMPs, receptor activator of nuclear factor-κβ ligand [RANKL]). Table 9-2 presents these biological mediators elicited by different MAMPs in resident and nonresident cells involved in the pathogenesis of destructive periodontal disease. As a result of the sheer proportion of fibroblasts in the periodontal tissues and also of the proximity and relevance of both fibroblasts and osteoblasts to nonmineralized and mineralized tissue turnover, respectively, these cells can play important roles in innate immunity during periodontal diseases. Epithelial cells represent the initial point of contact with MAMPs in the periodontium, and they play an important role in innate immunity not only because of their function as a physical barrier but also because they are equipped with PRRs and respond to MAMPs by secreting various cytokines and chemokines, including interleukin-8 (IL-8) and antimicrobial peptides (i.e., β-defensins, cathelicidins).173,73
TABLE 9-2
Biological Mediators Elicited by Different Microbial-Associated Molecular Patterns in Resident and Nonresident Cells Involved in the Pathogenesis of Destructive Periodontal Disease
| Cell Types | Pathogen-Associated Molecular Patterns | Biological Mediators | References |
| Epithelial cells | LPS, fimbriae, glycoprotein, whole bacteria, cell wall extracts, P. gingivalis gingipains | IL-8, G-CSF, GM-CSF, β-defensin-2, MMP-3, MMP-9, MMP-13, MIP-1α, IL-1β | 59,78,127,141,163,164,255,262,305,311,338,343,370 |
| Dendritic cells | Fimbriae, LPS, CpG DNA, DNA | IFN-α, IL-6, IL-8, IL-10, IL-12, TNF-α, GM-CSF | 172,183,184,283,335 |
| Endothelial cells | LPS, heat shock proteins | IL-6, GM-CSF, ICAM-1 | 77,103,180,224 |
| Gingival fibroblasts | LPS, peptidoglycans, CpG DNA, gingipains, T. forsythia detaching factor | IL-1β, IL-6, IL-8, TNF-α, PGE2, MCP-1, MMP-2 | 13,39,89,149,251,255,259,261,289,346,354,370,371 |
| Periodontal ligament fibroblasts | LPS | IL-6, IL-8, MMP-13, RANKL | 149,299,300,376 |
| Cementoblasts | LPS | OPN, OCN, RANKL, IL-6 | 244,249 |
| Macrophages | LPS, CpG DNA, leukotoxin | IL-1α, IL-1β, IL-6, IL-12, TNF-α, MMP-1, NO | 191,195,234,251,259,348,374 |
| Osteoblasts | LPS | IL-1β, IL-6, TNF-α, RANKL, PGE2, NO, MMP-2, MMP-9 | 194,274,339,372–375,393 |
| Neutrophils | DNA, LPS | IL-8, chemotaxis, shedding of L-selectin, MIP-1α | 305,355,356 |
| Monocytes | LPS, CpG DNA, fimbriae | IFN-γ, IL-1α, IL-1β, IL-6, IL-8, IL-12, TNF-α, LIF, RANKL, PGE2 | 27,28,40,77,87,103,129,144,175,206,222,243,289,293 |
| B lymphocytes | CpG DNA, heat stress proteins, cell sonicate extracts | IL-6, IL-10, IL-12, TNF-α, proliferation, antibody production | 54,258,367,389 |
| T lymphocytes | LPS, CpG DNA, peptidoglycan | IFN-γ, IL-4, IL-10, IL-13, inhibition of apoptosis, decrease in Th2 cytokines | 112,228,229,283,347,389 |
According to the current paradigm of the microbial etiology of periodontal disease, disease initiation depends on shifts in the microbial population of the dental biofilm toward a more complex flora that includes gram-negative and anaerobic species. This suggests that some bacterial species in the dental biofilm may not play a role in periodontal disease and that, in fact, periodontal clinical health is frequently observed despite the presence of a dental biofilm. However, the fact that different species of bacteria share common MAMPs (e.g., CpG DNA, lipopolysaccharides, peptidoglycans) that can potentially trigger an innate immune response suggests that tolerance mechanisms may counterbalance the vigilance aspect of innate immune responses to allow for the presence of these commensal, nonpathogenic bacteria. Tolerance mechanisms have been extensively studied in the gut epithelium, because the intestinal mucosa is continuously exposed to innocuous environmental antigens and commensal microorganisms that live in symbiosis with the host (for a review, see Reference 15). The critical importance of these tolerance mechanisms is exemplified by the fact that their dysregulation is associated with the pathogenesis of various inflammatory conditions, including inflammatory bowel disease and intestinal cancer.43,187
There are some important differences between the oral epithelium and the intestinal mucosa, however, especially the presence of specialized immune cells subjacent to the mucosal epithelium in the intestines (i.e., gut-associated lymphoid tissues and Peyer’s patches. However, the concept of a tolerance mechanism that renders the immune system hyporesponsive to commensal bacteria while retaining the capacity to respond to pathogenic organisms has also been investigated in oral epithelial cells. A comparison of the responses of oral epithelial cells to non-periodontopathic and periodontopathogenic bacteria demonstrated that, in general, commensal bacteria (e.g., Streptococcus gordonii, Streptococcus sanguinis) induce the expression of antimicrobial peptides without inducing the chemoattractant cytokine IL-8, whereas periodontopathogenic bacteria from the “orange complex” (i.e., Fusobacterium nucleatum and Prevotella intermedia) induce the strong expression of both antimicrobial peptides and IL-8. Interestingly, the bacteria that are more strongly associated with periodontitis (i.e., the “red complex” organisms Treponema denticola, Tannerella forsythia, and Porphyromonas gingivalis) tended to suppress the innate immune response by inhibiting the expression of antimicrobial peptides, of IL-8, or of both simultaneously.173,202,364 Tolerance mechanisms may involve the activation of different signaling pathways, because the activation of nuclear factor-κβ (NF-κβ; associated with the expression of many inflammatory genes) was not required for the expression of antimicrobial peptides by oral epithelial cells after stimulation with commensal bacteria as opposed to the periodontopathogenic microorganisms.59 There is still much to be explored in these tolerance mechanisms, but it is conceivable that commensal bacteria may be beneficial to the host, because they induce expression of antimicrobial peptides, which will clear some of the non-pathogenic and pathogenic bacteria while allowing non-pathogenic bacteria that are resistant to antimicrobial peptides (e.g., S. gordonii, S. mutans173,285) to establish themselves in the oral biofilm. The weak induction of IL-8 may function to maintain immune vigilance without causing too much collateral damage to the host tissues. On the other hand, pathogenic bacteria (e.g., the orange complex microorganisms, F. nucleatum and P. intermedia) that induce both antimicrobial peptides and IL-8 expression may be self-limiting their survival in the biofilm. Interestingly, the inhibition of the expression of antimicrobial peptides and IL-8 in epithelial cells by red complex bacteria (e.g., P. gingivalis, T. denticola) can represent host-evading strategies that allow them to survive and establish themselves in the oral biofilm.48
Defensins.
Antimicrobial peptides are components of the innate immune response in eukaryotes, providing defense against a wide spectrum of gram-positive and gram-negative bacteria, viruses, and fungi.142,372,391 In the oral cavity, at least 45 different antimicrobial peptides belonging to different biochemical classes are found in the saliva and the gingival crevicular fluid.119 Defensins and the cathelicidin LL-37 are the most studied antimicrobial peptides (for comprehensive reviews of various antimicrobial peptides, the reader is referred to References 119 and 177). Defensins and LL-37 are cationic peptides that bind to negatively charged molecules on the microbial cell surface (e.g., LPS in gram-negative bacteria and lipoteichoic acid in gram-positive bacteria), thereby depolarizing and permeabilizing the cell membrane and leading to bacterial cell death.199,322
Defensins are the predominant type of antimicrobial peptides in humans, and, on the basis of the spacing and pairing of cysteine residues, they can be subdivided into α- and β-defensins. α-Defensins can be further subdivided into those produced by polymorphonuclear neutrophils (also called human neutrophil peptides 1 through 4), which are most predominant in infection sites, and those produced by the Paneth cells of the small intestine (HD5 and 6). β-Defensins were initially identified in upper respiratory mucosal epithelial cells 20 years ago,85 and they are now known to be produced by a variety of epithelial cells, including cells from the skin, the upper respiratory tract mucosa, the kidneys, the pancreas, the lung, the uterus, the eye, and the urinary tract.75
The presence of defensins in the oral cavity of humans was first reported in 1995,320 and they have since been confirmed by various studies describing the abundant production of human β-defensins by the tissues in the oral cavity and by salivary glands. These defensins—particularly the β-defensins 1, 2, and 3—can be found in the saliva and the gingival crevicular fluid.75,91,197,277,306
On the periodontium, β-defensins are located in different regions of the epithelium: β-defensins 1 and 2 are observed in the upper layers of the gingival and sulcular epithelium, adjacent to the microbial biofilm and external environment, which is consistent with the innate immune “barrier” function of the epithelium. Interestingly, neither β-defensin 1 nor β-defensin 2 are found in the junctional epithelium. Protection in the junctional epithelium may be provided by the higher concentration of α-defensins and LL-37 produced by granulocytes migrating toward the gingival sulcus.75,74,233
The expression of defensins induced by whole periodontopathogenic bacteria such as F. nucleatum, P. gingivalis, A. actinomycetemcomitans, and T. denticola is largely dependent on TLR signaling, as various studies have demonstrated by silencing TLR expression in epithelial cell lines.174,278,328,364 For further information about the induction of defensins by periodontopathic bacteria, the reader is referred to Reference 118.
In addition to their primary antimicrobial function, defensins are modulated by immune response mediators and also present immunomodulatory functions of their own, which suggests that these peptides may actually function as an enhancer of innate immune response and even as a bridge between innate and adaptive immunity. Evidence that β-defensins 2, 3, and 4 induce the production of IL-6, IL-10, IL-18, IP-10, MCP1, MIP3, and RANTES by epithelial cells supports the idea of this immunoregulatory function of defensins.248,247 On the other hand, defensins are also modulated by soluble mediators secreted by immune cells, such as proinflammatory cytokines IL-1β, tumor necrosis factor-α (TNF-α), interferon-γ, and IL-17, which induce the expression of β-defensins 1, 2, and 3 by epithelial cells182,186,364; anti-inflammatory IL-4 and IL-10 suppress their production.186 Defensins also have a chemotactic effect on immune cells. The mechanisms are not fully understood and may involve direct interaction with unknown receptors in the target cells as well as indirectly inducing the production of chemokines. β-Defensins exert a chemotactic effect on macrophages, immature dendritic cells, memory T cells, and mastocytes via CCR6.384 Neutrophil-derived α-defensins are also involved in the chemokine receptor 6 of naive T cells and immature dendritic cells as well as the degranulation of mastocytes and complement activation.382,383
In the periodontium, the expression of β-defensins 1, 2, and 3 is observed at the mRNA level both in clinically healthy and diseased tissues; the expression of these epithelium-derived peptides appears to be correlated with periodontal health, thereby suggesting a protective role.38,46,365 On the other hand, the expression of neutrophil-derived defensins (α-defensins 1, 2, and 3) and LL-37 was significantly elevated in the gingival crevicular fluid of patients with chronic periodontitis282,357; however, the role of defensins and LL-37 in periodontal disease is currently not clear.
These antimicrobial and immunomodulatory roles of defensins have obvious attractiveness for therapeutic applications. However, the biochemical purification process is cost-inefficient and the synthesis process is complicated by the size and tridimensional structure of the peptides. Recently, novel analogs of defensins have shown even higher antibacterial activity than the endogenous β-defensins 1 and 3, without any cytotoxic effects on host cells,321 thus indicating the promise of this approach. The issues with the direct use of antimicrobial peptides with therapeutic purposes may also be dealt with by stimulating the endogenous production of these peptides.
Activation of the innate immune system is critical for lymphocyte activation and other immune cells to help clear the infectious microorganisms. However, the exuberant production of proinflammatory cytokines leads to severe pathology, including periodontal bone loss.18,81,82,122,123 To help prevent the deleterious effects of TLR activation, a number of signaling mechanisms have evolved. These mechanisms include the downregulation of surface TLR receptor expression; the transcriptional induction of negative regulators such as IL-1 receptor-associated kinase; the suppression of cytokine signaling-1 and SH2-containing inositol phosphatase; and the production of anti-inflammatory cytokines, including IL-10 and transforming growth factor-β (TGF-β).216 In contrast with the release of proinflammatory cytokines and mediators, the production of these negative immune regulators occurs over a much longer time frame, thereby providing a vital role for controlling the extent of pro- and anti-inflammatory mediators in a proper temporal sequence.216
Cell Signaling Pathways and the Expression of Biologically Active Mediators in the Innate Immune Response
TLRs are single-pass transmembrane proteins with N-terminals that present leucine-rich repeats that are responsible for the recognition of their ligands and with C-terminal cytoplasmic domains that are very similar to the cytoplasmic region of the interleukin-1 receptor,4 which are called Toll/IL-1 receptor domains (TIR domains). Thus, subsequent to the recognition of a ligand by TLRs, the signal generated makes use of pathways similar to those used by the IL-1 receptor. TLR signaling was originally described in the context of the activation of interferon regulatory factors (IRF) family of transcription factors and NF-κβ, thereby leading to the expression of interferon-γ and early-response inflammatory genes, respectively.
Interestingly, the participation of at least four adaptor proteins (MyD88, TRIF, Mal/TIRAP, and TRAM) that contain TIR domains and that can be recruited by activated TLRs results in important branching of the signal transduction and yields a significant flexibility to TLR signaling by allowing for cross-talk with other pathways, most notably the mitogen-activated protein kinase (MAPK) pathway. These adaptor proteins are recruited to TLRs by homophilic interactions between their TIR domains, and they are used differently by the TLRs. TLR5, TLR7, and TLR9 were shown to depend on the recruitment of MyD88 to signal,152,159 whereas TLR3 is the only TLR that does not use MyD88.379 TLR4, on the other hand, can use all four adaptor proteins: MyD88, TRIF, Mal/TIRAP, and TRAM.378–380 Although the activation of the canonical NF-κβ pathway is usually affected by all TLRs, the timing of NF-κβ activation188,190 as well as the additional signaling pathways that are activated by the branching of the signal varies among TLR receptors and with the participation of different adaptor proteins (Figure 9-2). These variations will ultimately affect the biological result in terms of gene expression, as demonstrated by the finding that, even though NF-κβ activation is observed after TLR4 stimulation by LPS, this may or may not result in inflammatory gene expression, depending on the adaptor protein used. In wild-type cells, LPS stimulation results in inflammatory cytokine expression, whereas, in MyD88-deficient cells, LPS fails to induce cytokine expression. In the absence of MyD88, the activation of NF-κβ occurs with delayed kinetics as compared with wild-type cells.188
The shift of the microbial population present in the oral biofilm from predominantly gram-positive to gram-negative bacteria that is associated with the onset of periodontal disease may lead to different patterns of immune response as a result of the type of TLR predominantly activated. Gram-positive bacteria were shown to activate TLR2, which induced increased expression of IL-8, whereas gram-negative bacteria activated predominantly TLR4, thereby resulting in the increased expression of TNF-α.351
However, some gram-negative microorganisms that are present in the oral biofilm and associated with periodontal disease are rather unique in their capacity to activate NF-κβ via the preferential use of TLR2.49 It has been reported that most gram-negative bacteria associated with periodontal disease—including Porphyromonas gingivalis, Tannerella forsythensis, Prevotella intermedia, Prevotella nigrescens, Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, and Veillonella parvula—are all capable of activating TLR2, whereas the latter two microorganisms also activate TLR4.193 Even though all of these disease-associated microorganisms activate TLR2 signaling, this pathway can also be activated in vitro by microorganisms that are present in an oral biofilm composed primarily of gram-positive bacteria, which are common colonizers of the oral biofilm and not associated with clinical signs of periodontal disease.388 The fact that TLR2 is activated by both pathogenic and non-pathogenic microorganisms is an interesting finding and suggests differences in the use of adaptor proteins as well as the concomitant activation of other TLRs by different MAMPs expressed by the various bacterial species that are present in an oral biofilm associated with disease. These differences can lead to the activation of different signaling pathways and the subsequent modulation of the host response.
NOD-like receptors include 23 genes in humans, even though some of these genes are primarily expressed in macrophages and polymorphonuclear leukocytes; the best studied and best characterized members, Nod1 and Nod2, are expressed by a wide variety of cells, including epithelial cells,358,360 monocytes/macrophages,260 and dendritic cells345 (see Table 9-1). The exact mechanism by which these proteins recognize their ligands is still unknown, and there is no evidence for direct interaction between ligands and NOD proteins. According to the proposed mechanism of activation of NOD proteins, they are kept in an inactive state via intramolecular interactions among the C-terminal leucine-rich repeat domain, the N-terminal fragment caspase-activating recruitment domain, and the NOD domains. Ligand recognition results in conformational changes that relieve the autoinhibitory intramolecular interactions and that allow for NOD-domain dependent nucleotide binding and oligomerization.162 The stimulation of Nod1 and Nod2 will result in the activation of NF-κβ and MAPKs, which is similar to the activation of TLRs (see Figure 9-2); however, the signal transduction pathways require different signaling intermediates. The activation of Nod2 leads to the recruitment of a kinase called receptor-interacting protein 2 (Rip2, also known as RICK), which will bind directly to the inhibitor of NF-κβ kinase-γ (IKKγ; also known as NF-κβ essential modulator or NEMO) and promote its ubiquitination and the activation of the catalytic regulatory subunits of the inhibitor of the IKK complex. Once activated, IKK phosphorylates the inhibitor Iκβ, thereby leading to its degradation by the proteasome, releasing NF-κβ, and allowing for its translocation to the nucleus, where it will affect the expression of various inflammatory genes.150,265 In this pathway, Rip2 has been shown to be required for Nod1 and Nod2 activation of NF-κβ,265 but, interestingly, its kinase activity is not essential.147 The same signaling intermediate Rip2 has also been shown to be involved in Nod1- and Nod2-induced MAPK activation by mediating the recruitment of the upstream activator of MAPK, TGF-β–activated kinase 1 (TAK1).147 However, although Rip2 and TAK1 are required for Nod1- and Nod2-mediated activation of MAPKs, the intermediate steps in the activation of this pathway are not well known.265,315
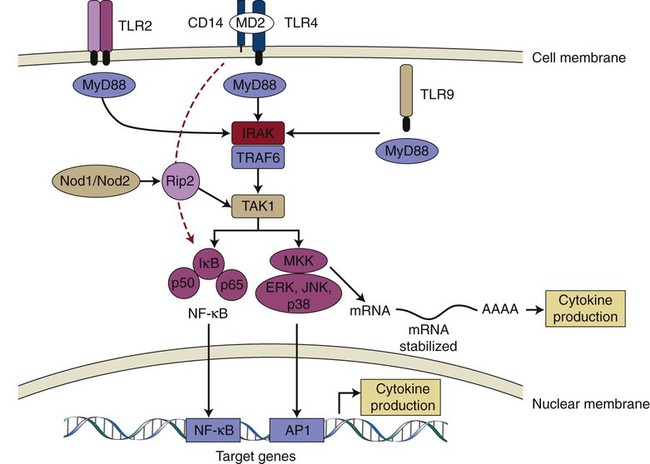
TLR-2, TLR-4, and TLR-9 are depicted as examples of TLR receptors expressed in cells of the periodontal tissues. After ligand binding, all TLRs (except TLR3) recruit adaptor protein MyD88 and activate the common upstream activator (IRAK/TRAF6 and TAK1) of NF-κβ and MAP kinases. TLR-4 may also activate NF-κβ independently of MyD88, with delayed kinetics (red dashed arrow). Nod1/Nod2 are cytosolic PRRs that recognize peptidoglycan fragments of the bacterial wall, and they may amplify the TLR-induced activation of signaling pathways. Activated NF-κβ and MAP kinases translocate to the nucleus and bind to their motifs (NF-κβ and AP-1, respectively) in the promoters of target genes (including early response and inflammatory genes) and induce their transcription into mRNA, which will ultimately lead to increased cytokine production. p38 MAP kinase is also involved after the transcriptional regulation of proinflammatory genes (e.g., IL-6, Cox-2) via the modulation of mRNA stability in the cytoplasm. TLR; Toll-like receptor; CD14, cluster of differentiation 14 molecule; MD2, myeloid differentiation protein-2; MyD88, myeloid differentiation primary response gene 88; IRAK, interleukin-1 receptor-associated kinase; TRAF6, tumor necrosis factor receptor associated factor-6; TAK1, transforming growth factor-β activated kinase 1; MKK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; AP-1, activator protein-1.
Because TLRs and NOD proteins are PRRs involved in the recognition of bacteria and considering that the signals generated by their activation converge to the same signaling pathways, there may be a synergistic or “amplifying” effect for simultaneous NOD and TLR activation by MAMPs. This cooperation between different PRRs in the activation of NF-κβ and MAPK signaling pathways may increase the sensitivity and potency of the host response to the presence of bacteria.98,99,363 Indeed, there is evidence that demonstrates that the activation of Nod1 and Nod2 has synergistic effects with TLR signaling on the production of proinflammatory cytokines, including IL-1, IL-4, IL-6, IL-10, IL-12, granulocyte macrophage colony-stimulating factor, and TNF.99,265,345,363 Moreover, it has been suggested that Nod1 and Nod2 signaling augments Th1 polarization induced by TLR signaling, except for TLR2 signals, which inhibit Th1-type cytokines as a result of IL-10 production.345
In addition to their role as PRRs and their synergism with TLR signaling, other members of the NOD family of proteins can also affect innate immune response via their role in the formation of the inflammasome, which is a multi-protein complex that activates caspase-1. Activated caspase-1 will process the inactive pro-IL-1β and pro-IL-18 forms that are produced and convert them into the biologically active forms that will be secreted.207
Inflammasomes.
The NLR proteins represent the “core” of the multi-protein complex, and they are reflected in the name of the inflammasome. NLRP1, NLRP3, and NLRC4 are the only inflammasomes with known physiological functions (Figure 9-3).273 In addition to the core NLR protein, inflammasomes also include caspase-1 and the apoptosis-associated speck-like protein, which contain caspase-activating recruitment domains as central proteins. The relevance of inflammasomes to the immune response is supported by the associations between mutations in the genes that encoding the proteins of the inflammasome as well as inflammatory autoimmune conditions, including vitiligo, Muckle-Wells syndrome, cryopyrin-associated periodic syndrome, and type 2 diabetes.179,156,210
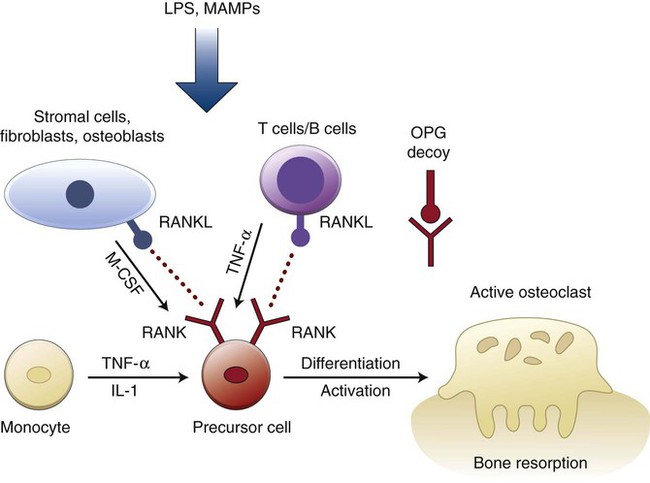
Schematic representation of the possible pathways leading to the activation of NLRP3 and NLRC4 inflammasomes. NLRP3 may be activated by a plethora of bacterial-derived stimuli, whereas NLRC4 is classically activated by microorganisms with type III or IV secretion systems, which are not usually associated with periodontal disease. However, other microorganisms as well as cytokines and biological mediators derived from the host may function as noncanonical activation pathways for both NLRC4 and NLRP3. In fact, bacterial infection can trigger the activation of several inflammasomes. Upon activation, the multiprotein complex recruits the adaptor protein ASC, which ultimately results in the activation of caspase-1 as the common effector molecule. Alternatively, ASC may also activate other signaling pathways (e.g., MAP kinases, NF-κβ) independently of the inflammasome, and this activation may also have an impact on the destruction of mineralized and nonmineralized tissues by modulating the expression of important biological mediators. Caspase-1 is the main effector protein of the inflammasomes, and it is responsible for the final processing of the inflammatory cytokines IL-1β, IL-18, and IL-33, which are not only relevant for the destruction of mineralized and nonmineralized tissues but which also can further activate signaling pathways that are important for the expression of other cytokines, inflammatory mediators, and protein constituents of the inflammasome itself in an autoregulatory positive feedback loop that amplifies the response. Active caspase-1 can also induce cell death by pyroptosis, which may reduce the number of viable osteoblasts in the periodontal microenvironment and aggravate the imbalance of the bone turnover.
The different inflammasomes are “assembled” and activated in response to various exogenous and endogenous signals. MAMPs and DAMPs may activate NLRP3, whereas NLRC4 is preferentially activated by MAMPs, including double-stranded DNA and specific bacterial proteins. The protease caspase-1 is the effector protein of the inflammasomes; it plays an important role in cell death (specifically by pyroptosis) and also in the final processing and activation of the inflammatory cytokines IL-1β, IL-18, and IL-33. Thus, the gain of function caused by constitutive or prolonged activation of the inflammasomes and consequently of caspase-1 may lead to an exacerbation of the inflammatory response by increasing the bioavailability of active IL-1β. On the other hand, loss or reduced function of the inflammasomes can dampen inflammation and attenuate the immune response, thereby reducing the ability of the host to detect and respond to various MAMPs. Various microorganisms (e.g., Salmonella typhimurium) present evasion mechanisms to prevent the activation of the inflammasomes and to facilitate their invasion, thereby indicating the relevance of inflammasomes for the immune response and host–microbial interactions.241
With periodontal diseases, there is scarce evidence of the importance of inflammasomes. There is an increase in the gene expression of NLRP3 and its endogenous antagonist NLRP2 in human gingival tissues with various forms periodontal disease as compared with gingival biopsies of periodontally healthy sites. Moreover, the expression levels were positively correlated with the levels of IL-1 and IL-18 in the tissues,41 thereby suggesting a role for NLRP3 inflammasome in inflammatory periodontal diseases. In vitro evidence indicates that microorganisms from the dental biofilm may modulate the expression of NLRP3 inflammasome and that this modulation was correlated with the production of IL-1 and IL-18.42 Interestingly, some periodontopathogens from the subgingival biofilm may evade immune surveillance by avoiding the activation of inflammasomes, as was demonstrated in vitro for P. gingivalis that attenuated NLRP3 and IL-1 gene expression by gingival fibroblasts.31
The production of cytokines is crucial for the establishment of a competent innate immune response and also for the subsequent activation of adaptive immunity. Cytokines such as IL-1, IL-18, and IL-33 are processed by the inflammasomes, and they play an important role in the polarization of T-cell response toward Th17, Th1, and Th2, respectively.60,209 Thus, inflammasome activation may bridge innate and adaptive immunity. An indication of the role played by inflammasomes on adaptive immunity comes from the experimental models of respiratory allergy and encephalitis. In these models, mice that are deficient in NLRP3, caspase-activating recruitment domains, or caspase-1 failed to develop an allergic reaction97,200 and showed reduced progression of encephalitis125,326 as a result of a marked shift in the adaptive response. Further evidence for the role of inflammasomes in the modulation of adaptive immunity includes models of host–microbial interactions that indicate a higher susceptibility for disseminated fungal infection in caspase-1–deficient animals; this is associated with the reduced Th1 and Th17 polarization of T cells.362
Complement Systems.
In cases of periodontal disease, the host defense also is dependent on a functional complement system, which coordinates the recruitment and activation of inflammatory cells, bacterial opsonization, phagocytosis, and lysis.128,131 In addition, complement can amplify the TLR4-mediated inflammatory response toward bacterial LPS challenge (this was reviewed in Reference 291). The complement system is critical to the linking of innate and adaptive immunity by regulating the activation of both B cells and T cells, either directly or through effects on antigen-presenting cells.
The activation of the complement cascade involves the sequential activation and proteolytic cleavage of a series of serum proteins via three distinct mechanisms, namely the classical, lectin, and alternative (Figure 9-4).128,291 Classical pathway activation occurs in response to antigen–antibody complexes that are recognized by the C1q subunit of C1. However, the lectin pathway is triggered through the interaction of a secreted pattern-recognition receptor (mannose-binding lectin) with specific carbohydrate groups on the surface of a variety of microorganisms. Both the classical and the lectin pathways then proceed through C4 and C2 cleavage for the generation of the classical/lectin C3 convertase (C4bC2b) (see Figure 9-4). The alternative pathway is initiated by the hydrolysis of C3 to C3(H2O), which is a C3b analog that forms the initial alternative pathway for C3 convertase, or through bacterial lipopolysaccharide and lipooligosaccharide molecules in concert with microbial-derived properdin.128 The alternative pathway also serves as a positive feedback loop for the classical and lectin pathways. All three pathways converge at the third component of complement (C3), which upon activation by pathway-specific C3 convertases leads to the generation of key effector molecules. These include the C3a and C5a anaphylatoxins, which activate specific G-protein–coupled receptors and which mediate the mobilization and activation of leukocytes. Also important are the C3b opsonins, which promote phagocytosis through complement receptors, and the C5b-9 membrane attack complex, which can lyse targeted pathogens (see Figure 9-4).128
All three pathways converge at the third component of complement (C3). The classical pathway is activated by antigen–antibody complexes, and it requires C1, C2, and C4 components. MBL activates the lectin pathway through MASPs and C2 and C4 cleavage. The alternative pathway is propagated through hydrolyzed C3 by complexing with factor B (fB) and via the fB cleavage of factor D (fD). The alternative pathway can also be activated via bacterial LPS in a properdin-dependent manner. Downstream from C3, proteolytic cleavage generates C3a and C5a anaphylatoxins, which activate the receptors C3aR and C5aR. C5aR can also be activated via C5L2. C5b initiates the assembly of C5b-9 MAC, which can induce bacterial lysis. Therapeutic blockage is depicted with the C3 and C5 components. MBL, mannose-binding lectin; MASPs, MBL-associated serine proteases; C5l2, C5a receptor-like-2; MAC, membrane attack complex.
The complement systems have also been shown to provide a barrier against the spread of bacterial infections; to facilitate clotting mechanisms291; to mobilize hematopoietic stem cells and progenitor cells from the bone marrow; to help replenish new leukocytes211; and to activate the differentiation of specific T-cell subsets.220 The dysregulation of complement activities may lead to a failure to protect the host against pathogens and amplify inflammatory tissue damage (this was reviewed in Reference 128). In the context of periodontal inflammation, complement subversion appears to play a major role in periodontal pathogenesis.132
Activated complement components are found in the gingival crevicular fluid of patients with chronic periodontitis as compared with healthy sites. Virtually all of the complement components have been detected in chronically inflamed gingiva or in the gingival crevicular fluid of patients as compared with healthy control samples. Finally, local complement activation may promote periodontal inflammation predominantly via C5a-induced vasodilation, increased vascular permeability and flow of inflammatory exudate, and chemotactic recruitment of inflammatory cells, especially neutrophils.128
Relative to periodontitis, therapeutic strategies are evolving that target either CR3 or CR5 (see Figure 9-4). Because C3 is a central component of all three activation pathways, blockade at this level is reasonable approach to treating complement-associated diseases, including periodontal diseases. CR3 antagonism through topical small molecule inhibitors has been shown to reduce P. gingivalis–induced alveolar bone loss.130,135 The complement-generated fragment C5a functions as a potent mediator of complement signaling and neutrophil recruitment that may protect but also mediate excessive neutrophil activation, and it has the potential to augment tissue damage during periodontal disease progression. As such, C5aR inhibitors have been recently used in preclinical models to indicate that the potential to inhibit C5aR may be a viable option for the treatment and management of periodontal diseases.1
Adaptive Immunity in Periodontal Diseases
Activation of Adaptive Immunity in Periodontal Diseases
In T lymphocytes, TLR agonists have been shown to modulate the expression of co-stimulatory receptors (e.g., CD28, CD69, and CD152) and to increase their proliferation and interferon-γ production, thereby suggesting that microbes and their MAMPs may also have a direct functional role in the regulation of adaptive immunity.51,331 This is especially important to realize in the periodontal disease context, which has been classically described as a chronic inflammatory condition and, as such, a condition that involves a dense lymphocytic infiltrate. Ever since the classical studies of the host response in periodontal disease—including the recognition of immunoglobulin-producing plasma cells in the gingival tissues of patients with periodontal disease47; the classical descriptions of initial, established, and advanced lesions264; and the characterization of the inflammatory infiltrate in the dog model217—the nature of the inflammatory infiltrate and its association with a certain type of inflammatory response and ultimately with the quiescence or progression of disease, as characterized by bone resorption, has been debated.
The maturation process induced by MAMPs that leads to the migration of DCs to the lymph nodes involves the modulation of the expression of various chemokine receptors that renders DCs less responsive to inflammatory signals and more responsive to lymphocyte-derived signals. Notably, the LPS-induced maturation of DCs has been shown to result in the downregulation of “inflammatory” CCR1 and CCR5 (which responds to macrophage inflammatory protein-1α, for example) and the upregulation of CXCR4 and CCR7 (which respond to CXCL12 and CCL19, respectively).309 These changes ultimately control DC trafficking and migration to the lymph nodes, and they represent one mechanism whereby the modulation of cell-signaling pathways (i.e., MAMPs activating PRR signaling versus inflammatory cytokine signaling) can influence the host response. The profound phenotypic changes that DC cells undergo during the process of activation by MAMPs and conversion into antigen-presenting cells is a multi-step process that includes not only trafficking and migration into lymph nodes but also the expression of high levels of major histocompatibility complex (MHC) bearing the processed microbial-derived peptides that will engage T-cell receptors in naïve T cells; the expression of co-stimulatory membrane-bound molecules (e.g., CD80, CD86), which are important for T-cell survival and proliferation; and finally the production of mediators (e.g., IL-12) that act on T cells and that induce their terminal differentiation into an effector cell type and their profile of cytokine expression. Ultimately, the integration of these signals (i.e., MHC antigens, co-stimulatory molecules, cytokines) will determine the fate and nature of the adaptive immune response.181
Interestingly, many of the features of DC activation can be induced by inflammatory cytokines in the absence of MAMPs and PRR signaling. This fact is interpreted by some as being supportive of the “danger model” of immunity, in which endogenous danger signals (e.g., toxic products from necrotic, apoptotic, or infected cells; inflammatory cytokines) can also trigger an immune response, similarly to exogenous MAMPs.30,231 There are two important arguments against this concept:
1. Inflammatory cytokines may be synergistic with MAMP signaling to induce DC maturation, because cytokines are produced by various innate immune cells in response to the PRR signaling triggered by MAMPs. This would turn the inflammatory (or “danger”) signals into “complementary” or surrogate signals of response to the MAMPs and not necessarily as alternatives.250
2. Activated or mature DCs defined by their high levels of expression of MHC class II molecules and co-stimulatory receptors CD80, CD86, and CD40 may not be fully competent for the activation of T-cell responses.5,100
The signaling aspects involved in DC maturation and activation are especially interesting, because most PRR and inflammatory receptor signals converge into similar pathways (e.g., NF-κβ, MAPKs). Despite similar signaling pathways activated by MAMPs and inflammatory cytokines, MyD88-defficient DCs stimulated with inflammatory cytokines fail to produce IL-12 and other inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-β),160 which will have important consequences for the differentiation and activation of naïve CD4+ and CD8+ T cells71 and also indirectly affect the activation of B cells and the humoral response.336 Among the possible reasons for this differential role of MAMPs and inflammatory cytokines as external signals, despite the activation of similar signaling pathways, are the kinetics of the activation of the signaling pathways and also the other signaling pathways that are simultaneously activated by each external signal. The integration of multiple signal pathways may be required for a given profile of cytokine expression by DCs. Specifically, in patients with periodontal diseases, both MAMPs and inflammatory cytokines are usually present to fully activate the DCs, which suggests that there is no impairment to a competent activation of adaptive immunity.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


