Dermatological Diseases
•. Acrodermatitis Enteropathica
•. Hailey–Hailey Disease (Familial Benign Chronic Pemphigus)
•. Darier’s Disease (Keratosis Follicularis)
•. Incontinentia Pigmenti (Bloch-Sulzberger Syndrome)
•. Kawasaki Disease (Mucocutaneous Lymph Node Syndrome)
•. Tuberous Sclerosis Complex (Epiloia, Bourneville’s Disease)
Lichen Planus
Clinical features
Oral LP almost invariably occurs as a bilateral disease and it involves the posterior buccal mucosa followed less commonly by the tongue, gingiva, hard palate, and the labial mucosa. Although any site can be involved, palatal and sublingual lesions are very uncommon.
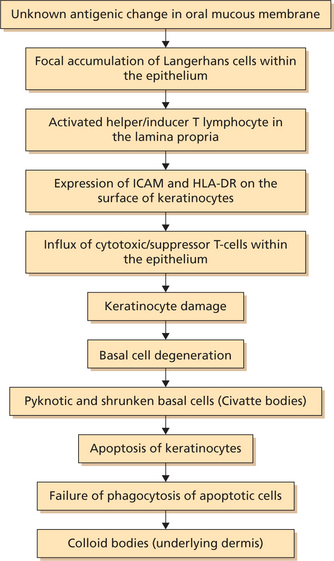
Clinically oral LP appears as radiating white or gray velvety thread like lesion, which consists of papules in linear, annular or retiform arrangement. A tiny white elevated dot is present at the intersection of the white lines known as ‘Wickham’s striae’ or ‘Honiton lace’.
An isomorphic response (Koebner’s phenomenon) is common occurrence in LP, and develops in areas previously subjected to some type of trauma (Figure 1). Reticular is the most common type, consisting of slightly raised fine whitish lines in an interlocking lace like keratotic pattern. Papular lesions are small (0.5–1.0 mm) white raised papules. Plaque type closely resembles leukoplakia with a reticular surrounding. Atrophic type appears as inflamed areas of mucosa covered by thin red appearing epithelium. Erosive type presents with atrophic mucosa with ulcers. Bullous form is very rare and is characterized by formation of large thin walled bullae.
Differential diagnosis
| Reticular form | – Lichenoid reactions |
| Plaque form | – Leukoplakia, hyperplastic candidiasis, traumatic keratosis |
| Atrophic form | – Speckled leukoplakia, anemic stomatitis, systematic lupus erythematosus and discord lupus erythematosus |
| Erosive and bullous form | – Vesiculobullous lesions |
| Annular form | – Erythema circinata migrans. |
Management
The role of Candida in the causation of oral LP has been debated, therefore a smear for candida needs to be made and if positive, a topical antifungal—clotrimazole (available as Candid gum paint in India) should be given for 14 days.
Treatment of oral lichen planus is given in Flowchart 2 on page no 153.
Epidermolysis Bullosa
Classification

 Epidermolysis bullosa simplex with muscular dystrophy
Epidermolysis bullosa simplex with muscular dystrophy
 Epidermolysis bullosa atrophicans genaralisata graves
Epidermolysis bullosa atrophicans genaralisata graves


The current classification proposed for epidermolysis bullosa is mentioned in Chapter 7 on Vesiculobullous Disorders.
Clinical features
Management
Although the data are preliminary, they suggest that intravenous immunoglobulins may be a promising treatment modality for resistant, non-responsive or refractory EB acquisita.
Psoriasis




Clinical features
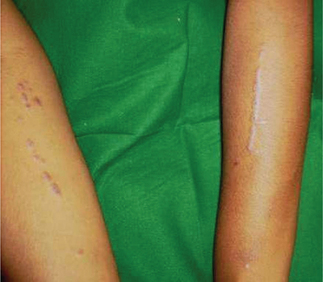
Psoriasis is more common in whites and in women. Approximately 10–15% of new cases begin in children younger than 10 years. The median age at onset is 28 years. Typical skin lesions of psoriasis appear as well-circumscribed erythematous patches with overlying thick silvery scales (Figure 2A, B). Lesions may occur in any location, but most commonly involve the scalp and the anterior hairline, torso, bony prominences of the extremities, nails, perianal and perineal areas. The course of the disease is unpredictable and characterized by spontaneous episodes and relapses.
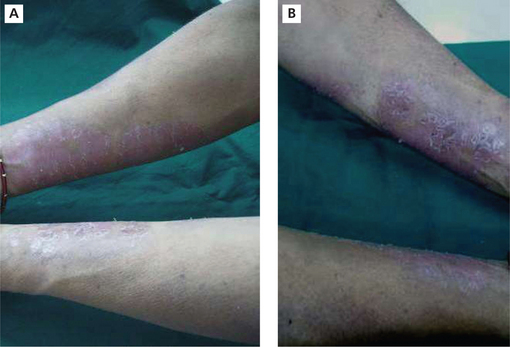
Figure 2 Erythematous patches on the forearms along with silvery scales in psoriasis. Courtesy: Department of Oral Medicine and Radiology, MCODS, Mangalore
If the deep scales are removed, one or more tiny bleeding points are disclosed, which is popularly referred to as Auspitz’s sign.
Van der Waal and Pindborg described four types of oral psoriatic lesions.




Ectodermal Dysplasia
Clinical features
The condition is characterized by hypodontia, hypotrichosis, and hypohidrosis. Neonates exhibit excessive scaling of the skin and unexplained pyrexia. Patients present with sparse hair and eyebrows (Figure 3A, B).
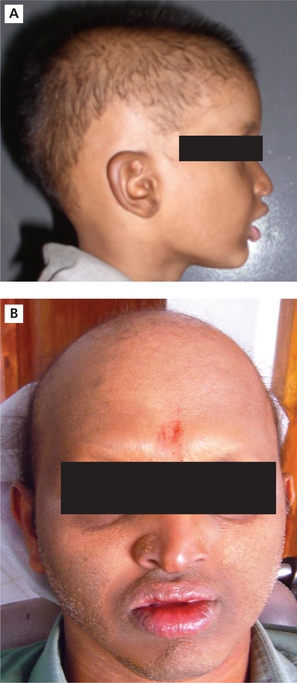
Figure 3 (A) A boy with sparse scalp hair in ectodermal dysplasia. Courtesy: Dr Sumanth KN. (B) Photograph showing hair loss of the eyebrows and scalp hair. Courtesy: Department of Oral Medicine and Radiology, MCODS, Mangalore
As the age progresses frontal bossing, saddle nose, sunken cheeks, thick/everted lips wrinkled and hyperpigmented skin around the eyes are seen. Patients may present with fever of unknown origin because of the inability to sweat.
Oral manifestations
Anodontia/oligodontia is often seen. The remaining teeth are usually malformed. Both deciduous and permanent teeth are affected. Most common missing teeth are molars. Malformed teeth have truncated and conical crowns and shortened roots (Figures 4 and 5).
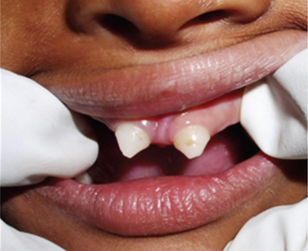
Figure 4 Hypoplastic and conical primary maxillary central incisors in ectodermal dysplasia. Courtesy: Dr Sumanth KN
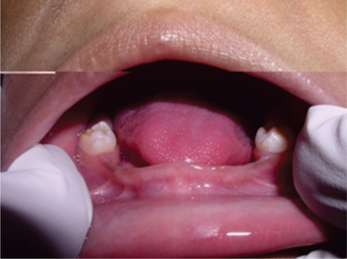
Figure 5 Hypoplastic and malformed mandibular second primary molars as well as underdeveloped alveolar ridges in ectodermal dysplasia. Courtesy: Dr Sumanth KN
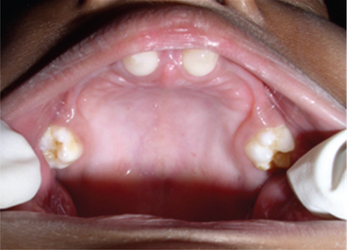
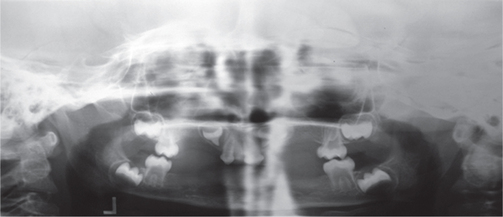
Dry mouth, high palatal arch and cleft palate may be seen in some individuals. The alveolar ridges are usually malformed (Figure 6). Orthopantomograph will help to confirm the absence of teeth (Figure 7).
Ehlers–danlos Syndrome
There are six major types of EDS that are characterized by distinctive features.
Villefranche classification of EDS
 Classical EDS (similar to Berlin Type I [gravis] and Berlin Type II [mitis])—Type V collagen defects.
Classical EDS (similar to Berlin Type I [gravis] and Berlin Type II [mitis])—Type V collagen defects.
 Hypermobility EDS (similar to Berlin Type III [hypermobile])—unknown collagen defects.
Hypermobility EDS (similar to Berlin Type III [hypermobile])—unknown collagen defects.
 Vascular EDS (similar to Berlin Type IV [arterialecchymotic])—Type III collagen defects.
Vascular EDS (similar to Berlin Type IV [arterialecchymotic])—Type III collagen defects.
 Kyphoscoliosis EDS (similar to Berlin Type VI [ocularscoliotic])—defect in lysyl hydroxylase.
Kyphoscoliosis EDS (similar to Berlin Type VI [ocularscoliotic])—defect in lysyl hydroxylase.


Clinical features











Oral manifestations
Radiographic examination often reveals pulp stones and roots that are short and deformed.







Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


