9
Bleeding Disorders
I. Background
Description of Disease/Condition
Bleeding disorders are conditions of varying severity that affect normal hemostasis, resulting in prolonged or excessive bleeding. These disorders may be congenital or acquired and cause quantitative and/or qualitative abnormalities in blood elements (i.e., vascular endothelial cells, platelets, coagulation proteins). Specific conditions are defined by deficiencies or abnormalities of blood components (see Table 9.1).
Table 9.1. Definitions of Bleeding Disorders
| Platelet disorders | |
| Thrombocytopenia | Decreased number of functioning platelets caused by decreased platelet production or accelerated platelet destruction/removal |
| Immune thrombocypenic purpura (ITP) | An autoimmune disorder causing platelet destruction due to the presence of antibodies against the patient’s own platelets |
| Drug-induced platelet disorders | Drugs may reversibly or irreversibly cause inhibition of platelet function. |
| Coagulation disorders | |
| von Willebrand disease | An autosomal dominant hereditary bleeding disorder caused by a deficient or defective plasma von Willebrand factor (vWF) |
| Hemophilia A | An X-linked genetic disorder resulting in deficient or defective clotting Factor VIII |
| Hemophilia B | An X-linked genetic disorder resulting in deficient or defective clotting Factor IX |
| Disseminated intravascular coagulation (DIC) | An acquired coagulation disorder characterized by uncontrolled thrombin activation and release, resulting in severe thrombosis that may be fatal |
| Drug-induced coagulation disorders | Drugs may prevent synthesis of coagulation cascade factors and have the potential to result in prolonged bleeding. |
Pathogenesis/Etiology
Hemostasis is the process of blood clot formation and can be divided into four phases: (1) vasoconstriction, (2) platelet plug formation, (3) blood coagulation, and (4) fibrinolysis. Immediately following tissue injury, damaged blood vessels constrict, which temporarily decreases blood flow and pressure within the vessel. Vasoconstriction is followed by mechanical blockage of the injury by a platelet plug. Platelets become activated and adhere to damaged endothelium to form a loose platelet plug. These steps initiate a series of reactions known as the coagulation cascade. These enzymatic reactions involving 13 coagulation factors end in the formation of a fibrin clot that stabilizes the platelet plug. Eventually, as the damaged vessel repairs itself, the clot is degraded proteolytically through a process called fibrinolysis.
Bleeding or bruising that is spontaneous or excessive after injury may be caused by abnormal platelet number/function or vascular integrity and/or defects in coagulation or fibrinolysis. Most bleeding disorders are caused by quantitative or qualitative abnormalities of platelets or coagulation proteins.
Platelet Disorders
Quantitative platelet defects can result from decreased production of platelets caused by bone marrow dysfunction, increased splenic sequestration, or increased consumption of platelets in acquired medical conditions (i.e., immune thrombocytopenic purpura [ITP]). ITP is characterized by the development of antibodies to one’s own platelets. Thrombocytopenia can also develop from dysfunctional platelet activity, which may be due to medications or acquired medical conditions (i.e., hematological malignancies, bone marrow disorders, end-stage renal disease). There are also a number of rare inherited qualitative platelet disorders that can result in symptoms from mild bleeding to severe mucocutaneous hemorrhage. Examples include Glanzmann thrombasthenia, Bernard–Soulier syndrome, and platelet storage pool defects.1
Coagulation Disorders
Coagulation disorders may be congenital or acquired. Congenital coagulation disorders are uncommon and are characterized by the presence of a single abnormality that can account for the entire clinical picture. von Willebrand disease (vWD) is an autosomal dominant condition that results from the deficiency of von Willebrand factor (vWF) associated with Factor VIII, which varies in severity and type. Many individuals with vWD remain undiagnosed until exposed to trauma or surgery. Hemophilias are sex-linked recessive disorders that result in a deficiency of Factor VIII (in hemophilia A) or Factor IX (in hemophilia B). Although almost all hemophiliac patients are male, it is possible for females who are usually carriers to be affected. Patients with hemophilia A or B often present with similar clinical presentations.
The clinical severity of hemophilia is typically inversely proportional to the measured level of factor coagulant activity (e.g., Factor VIII level). Factor levels are genetically determined and do not vary with time. Severe disease occurs at factor level of less than 1% of normal, moderate at 1–5%, and mild disease above 6%. Normal coagulation factor levels range from 60% to 150%. Approximately 60% of all cases of hemophilia A are severe, while 20–45% of hemophilia B cases are severe. Inhibitors or alloantibodies to Factor VIII and Factor IX develop in 10–30% of patients with severe hemophilia due to genetic and environmental exposures. Factor VIII inhibitor levels are assessed with the Bethesda inhibitor assay (BIA) where the number of Bethesda units (BUs) reflects the inhibitor level, and the amnestic response of the inhibitor to factor concentration replacement therapy reflects the response type. A high-titer, high-responder-type inhibitor patient is the most difficult to manage with factor replacement products because normal replacement products are rapidly inactivated by the inhibitor becoming ineffective.
Acquired coagulation disorders are more common than congenital disorders and are more complex in their pathogenesis. Disseminated intravascular coagulation (DIC) is a condition in which massive activation of the clotting cascade gives rise to uncontrolled thrombin formation, resulting in thrombosis and further activation of the clotting cascade, causing further coagulopathy and bleeding. This may be a manifestation of underlying leukemia, other cancer, obstetric complications, massive tissue injury, or infections, and requires urgent management. Patients with severe liver disease are susceptible to hemorrhage because many of the proteins involved in the coagulation cascade are synthesized in the liver, particularly Factors II (thrombin), VII, IX, and X. Decreased levels of such factors are observed in patients with severe liver disease. In addition, there is reduced clearance of activated clotting factors by the liver. Furthermore, moderate thrombocytopenia is common, resulting from decreased platelet production and increased platelet destruction by hypersplenism.
Drug-Induced Disorders
Certain drugs can cause acquired platelet disorders. One of the most common drugs is aspirin, which irreversibly binds to the enzyme cyclooxygenase, causing blockage of thromboxane and prostaglandin synthesis, thereby preventing platelet release and aggregation for the life of the platelet (approximately 9 days). Other nonsteroidal anti-inflammatory drugs (NSAIDs) also inhibit cyclooxygenase, but the inhibition is reversible. Dipyridamole (Persantine® or combined with aspirin in the drug Aggrenox®) is a thromboxane synthase inhibitor that blocks platelet aggregation. Platelet adenosine diphosphate (ADP) receptor agonists, such as toclopidine (Ticlid®), clopidogrel (Plavix®), and prasurgel (Effient®), are prescribed for the prevention of thromboembolic disease and atherosclerotic events. These antiplatelet agents inhibit platelet aggregation by interfering with platelet ADP binding and are irreversible inhibitors of platelet function. It generally takes 5–7 days after discontinuation of irreversible platelet inhibitors for platelets to return to their baseline function.
Drug-induced coagulation disorders are most commonly caused by the coumarin anticoagulants such as warfarin (Coumadin®), which is prescribed to prevent venous thrombosis and systemic embolism in susceptible patients. Coumadin® acts as a vitamin K antimetabolite, interfering with the synthesis of Factors VII, IX, and X as well as prothrombin. The primary effect of Coumadin® is in the common pathway due to inadequate amounts of prothrombin. In addition, heparin and low-molecular-weight heparins (LMWHs) are fast-acting anticoagulant drugs that bind antithrombin III, which inhibit thrombin and other coagulation factors, resulting in the inhibition of fibrin formation. Fondaparinux (Arixtra®) is an injectable Factor Xa inhibitor used similarly to the LMWH drugs. Newly available oral direct thrombin (Factor IIa) inhibitor dabigatran (Pradaxa®) and Factor Xa inhibitor rivaroxaban (Xarelto®) are used to prevent stroke in patients with atrial fibrillation and venous thromboembolism in patients who have recently undergone total knee or hip replacement. They do not require laboratory monitoring, have less associated bleeding complications than Coumadin®, and have half-lives of 14–17 hours (dabigatran) and 5–13 hours (rivaroxiban), compared with 2.5 days for warfarin.2
Epidemiology
There are numerous etiologies of platelet and coagulation abnormalities. More common congenital and acquired conditions are the following:
- ITP: Estimated incidence of 50–100 new cases per year, equally distributed between children and adults. ITP is more prevalent in young women.
- vWD: The most common inherited bleeding disorder in the United States with a prevalence estimated to be 1.3% of the general population.3
- Hemophilia A: Hemophilia A accounts for approximately 80% of hemophilia cases in the United States, affecting approximately 1 in 5000 male births.
- Hemophilia B: Hemophilia B accounts for approximately 13% of hemophilia cases in the United States, affecting approximately 1 in 35,000 male births.
- Coumarin Therapy-Induced Anticoagulation Disorder: Coumadin® is prescribed to approximately 1 million individuals in the United States each year.
 Coordination of Care between Dentist and Physician
Coordination of Care between Dentist and Physician
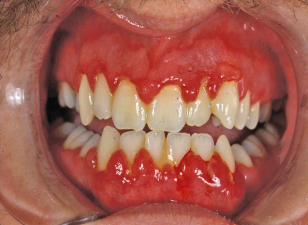
- The first step in assessing the risk for bleeding is obtaining a comprehensive medical and medication history. A bleeding disorder may be suspected when a patient complains of excessive bruising or bleeding that may be secondary to known trauma or bruising at sites with no recollection of trauma. Oral signs include gingival oozing (see Figs. 9.1 and 9.2), petechiae, and ecchymoses. In patients who are considered to be at risk for bleeding, appropriate laboratory tests should be performed and interpreted to evaluate the potential for bleeding complications. If warranted, prophylactic measures should be taken prior to performing dental care.
- Dental procedures that involve any type of soft or hard tissue damage can potentially cause bleeding. Patients with a diagnosed bleeding disorder require management in coordination with a medical provider prior to performing any dental procedure that has the potential to cause bleeding. Oral soft tissue lesions, including petechiae, ecchymoses, and hematomas, are frequently encountered in patients with clinically significant bleeding disorders.
- Depending on the underlying disorder and its severity, measures may have to be taken prior to, during, and following dental procedures. Consultation with a physician or specialist physician including hematologist or cardiologist is recommended in more complicated cases. In some circumstances, treatment may be best provided in a hospital setting, where pre- and/or posttreatment infusions and/or emergency management can be more easily coordinated.
Figure 9.1 Spontaneous gingival oozing in a patient with severe thrombocytopenia.
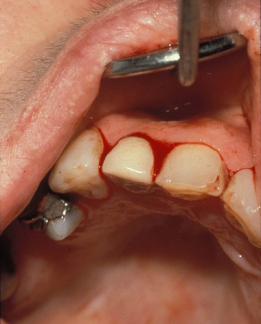
Figure 9.2 Spontaneous gingival oozing in a patient with leukemia and severe thrombocytopenia.
 II. Medical Management
II. Medical Management
Identification
Bleeding disorders vary in severity and etiology. Many patients with congenital and/or severe bleeding disorders may be aware of their underlying condition. However, patients with newly acquired disease may be unaware of their disorder. Furthermore, patients on antiplatelet or anticoagulant medications that may contribute to abnormal bleeding tendencies may not be aware of their increased risk for bleeding. Bleeding symptoms differ based on the origin of the hemostatic disorder, and clinical presentation can assist in establishing the diagnosis. See Table 9.2.
Table 9.2. Clinical Bleeding Symptoms Differ Based on Nature of Hemostatic Disorder
| Clinical Findings | Platelet and Vascular Disorders | Coagulation Disorders |
| Petechiae | Characteristic | Rare |
| Ecchymoses | Characteristic, usually small and multiple | Common, often large and solitary |
| Deep dissecting hematomas | Rare | Characteristic |
| Hemarthroses | Rare | Characteristic |
| Delayed surgical bleeding | Rare | Common |
| Bleeding from superficial cuts and scratches | Persistent, often profuse | Minimal |
Medical History
A thorough medical and medication history is critical prior to performing dental care on a patient to assess risk for bleeding. Furthermore, patients diagnosed with bleeding disorders prior to 1985 may have received contaminated blood transfusions and may be at risk for blood-transmitted infectious diseases, such as HIV/AIDS, hepatitis B, and hepatitis C. Affirmative responses to confidential medical history questions in the following areas will alert the dentist to the possible need for further inquiry:
- hemophilia, vWD, or other congenital coagulation disorders;
- bruises easily;
- epistaxis (frequent nosebleeds);
- prolonged bleeding after trauma/surgery including dental procedures;
- history of multiple blood transfusions;
- blood malignancies or dyscrasias;
- current or recently terminated cancer treatment;
- advanced liver disease/hepatitis;
- end-stage renal disease receiving hemodialysis (heparin is administered during dialysis treatment);
- current use of antiplatelet or anticoagulant medications such as aspirin, Coumadin®, ADP inhibitors, LMWH therapy, direct thrombin or Factor Xa inhibitors;
- medical conditions for which one may be on prophylactic antiplatelet or anticoagulant therapy:
- atrial fibrillation,
- prosthetic heart valve,
- history of deep vein thrombosis (DVT),
- post-myocardial infarction,
- history of cerebrovascular accident (CVA).
Physical Examination
The physical examination may contribute to />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


