Haematology
Key Points
• Postoperative bleeding usually has a local cause
• Von Willebrand disease is the most common inherited bleeding disorder
• Warfarin anticoagulation is common
Haemostasis
Normal haemostasis (bleeding cessation) depends on the interaction of blood vessels, platelets, fibrin coagulation and deposition, and fibrinolytic proteins (Fig. 8.1). Three reactions – primary, secondary and tertiary haemostasis – act simultaneously. Excessive blood clotting is prevented by the endothelium, which provides a physical barrier and secretes products that inhibit platelets (e.g. nitric oxide and prostaglandin I2 [prostacyclin]). Inhibitors limit haemostasis to the site of injury and prevent overproduction of the clot – which could lead to pathological thrombosis.
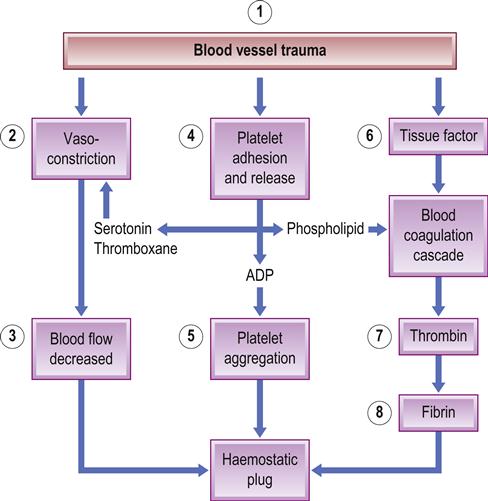
Primary Haemostasis (Vascular and Platelet Activity)
Primary haemostasis is by vasoconstriction after injury, which retards blood loss, slows local blood flow, enhances platelet adherence to subendothelial surfaces and activates coagulation.
The endothelial lining of the vessel wall is a barrier between the circulating platelets and the prothrombotic subendothelial matrix. Upon vessel injury, the endothelial layer is disrupted and the circulating platelets interact with the subendothelium through adhesive receptors, such as glycoprotein (GP) Ib-IX-V receptors, that mediate rolling and tethering of the platelets to von Willebrand factor (vWF) at the site. Platelet collagen receptors α2β1 and GP VI then mediate more firm adhesion and further platelet activation, causing release of platelet dense granules contents; these contain agonists such as adenosine diphosphate (ADP), and α-granules, which contain fibrinogen, factor V and P-selectin. The platelet generates lipid mediators such as thromboxane A2. ADP elicits its effects on the platelet through the P2Y1 and P2Y12 receptors, whereas thromboxane A2 activates the thromboxane-prostanoid (TP) receptor on the platelet surface. The release of the granule contents also triggers the blood coagulation response as a result of the release of clotting factor V, and the inflammatory response through the exposure of P-selectin on the platelet surface. Also, tissue factor is exposed, which initiates the coagulation response that results in formation of thrombin. Thrombin activates platelets via interactions with the proteinase-activated receptor-1 (PAR1) and PAR4 receptors and also cleaves fibrinogen to form fibrin. Fibrin further stabilizes the accumulating platelet plug at the site of injury, resulting in a stable haemostatic plug.
Platelet activation in response to vessel injury is thus important for the arrest of bleeding (Fig. 8.2), but activation during disease states leads to vascular occlusion and ischaemic damage. The P2Y12 receptor, activated by ADP, plays a central role in platelet activation and is the target of P2Y12 receptor therapeutic antagonists such as clopidogrel (antiplatelets).
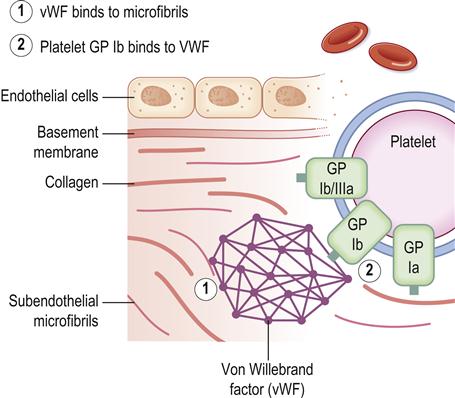
Abnormalities in primary haemostasis
Abnormalities in primary haemostasis (abnormal platelet number or function, abnormal vWF or defects in the blood vessel wall) lead to haemorrhage from mucosal surfaces (epistaxis, gingival bleeding, melaena, haematuria), and into skin and mucosae (petechial or ecchymotic haemorrhages). Prolonged bleeding from wounds is usually restricted by blood coagulation. However, if the defect is severe, more pronounced bleeding can result (e.g. intracavity haemorrhage).
Primary haemostasis inhibitors are the natural inhibitors of platelet function, such as bradykinin, prostacyclin and nitric oxide, released by endothelial cells. Acquired inhibitors of platelet function are rare, whereas acquired inhibitors of vWF (platelet GP Ib) form in a variety of diseases and result in acquired von Willebrand disease (avWD).
More commonly, platelet function is inhibited intentionally by the administration of agents such as aspirin or clopidogrel for the prevention of thrombosis.
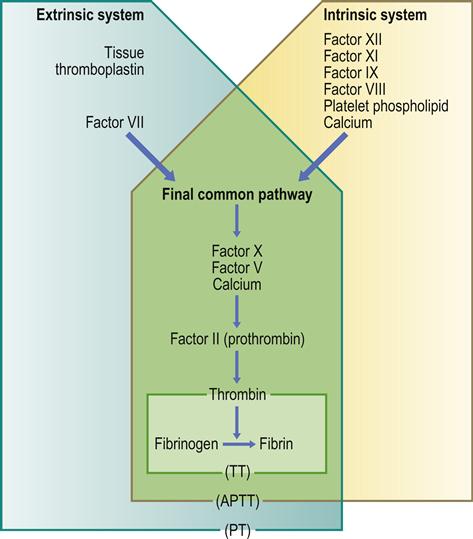
Secondary Haemostasis (Blood Coagulation or Clotting)
Secondary haemostasis is the formation of fibrin through the coagulation cascade (Table 8.1, Fig. 8.3), which is traditionally separated into three pathways (extrinsic, intrinsic and common) involving blood coagulation factors acting as enzymes, which require activation and cofactors.
Table 8.1
| Factor | Name |
| I | Fibrinogen |
| II | Prothrombin |
| III | Thromboplastin |
| IV | Calcium |
| V | Proaccelerin |
| VI | – |
| VII | Proconvertin |
| VIII | Antihaemophilic factor |
| IX | Christmas factor (plasma thromboplastin component) |
| X | Stuart–Prower factor |
| XI | Plasma thromboplastin antecedent |
| XII | Hageman factor |
| XIII | Fibrin-stabilizing factor |
| Fitzgerald factor | High-molecular-weight kininogen |
| Fletcher factor | Prekallikrein |
APTT=activated partial thromboplastin time; PT=prothrombin time; TT=thrombin time.
The extrinsic pathway is the main pathway. It is initiated by the generation/exposure of tissue factor (factor III), with expression being upregulated by cytokines (tumour necrosis factor alpha [TNFα], interleukin [IL]-6), and the tissue factor–factor VII complex, the latter activating factor X.
The intrinsic pathway amplifies coagulation, is stimulated by thrombin, through activation of factor XI, and involves high-molecular-weight kininogen, prekallikrein, factors XII, XI and IX, and factor VIII, which acts as a cofactor (with calcium and platelet phospholipid) for the factor IX-mediated activation of factor X. A minor route of stimulation of the intrinsic pathway (the alternative pathway) is the direct activation of factor IX by the tissue factor–factor VII complex.
The common pathway involves the factor X-mediated generation of thrombin from prothrombin (facilitated by factor V, calcium and platelet phospholipid [‘prothrombinase complex’]). Thrombin then activates factors XI and VIII, amplifying the coagulation cascade. Activated factor IX, together with activated factor VIII, calcium and phospholipid (‘tenase complex’), amplify the activation of factor X, generating large amounts of thrombin.
Thrombin cleaves fibrinogen to form soluble fibrin monomers, which spontaneously polymerize to soluble fibrin polymer; thrombin also activates factor XIII, which, together with calcium, cross-links and stabilizes the soluble fibrin polymer, resulting in cross-linked (insoluble) fibrin. Thrombin also promotes coagulation by promoting platelet aggregation, and activating factors V and VIII – necessary cofactors for the ‘prothrombinase’ and ‘tenase’ complexes, respectively. Thrombin also activates factor XIII, essential for the cross-linking of the fibrin polymer, and inhibits fibrinolysis by generation of a thrombin-activatable fibrinolytic inhibitor (TAFI).
Drugs that act as anticoagulants include warfarin, new oral anti-coagulants (NOACs), heparin and hirudin.
Abnormalities in the coagulation cascade
These cause more serious bleeding than do defects of primary haemostasis, resulting in bleeding into cavities (chest, joints and cranium) and subcutaneously (haematomas). Petechial haemorrhages are rare. The main coagulation inhibitor is antithrombin (AT; also called antithrombin III, or ATIII), a liver alpha2-globulin that inhibits thrombin (factor IIa) and many activated coagulation proteins (including factors II, IX, X, XI and XII). Heparin, produced in vivo by degranulated mast cells or basophils, enhances antithrombin binding to thrombin, and this is the basis for its use as an anticoagulant.
Protein C, a vitamin K-dependent protein produced in the liver, inactivates factors V and VIII. Protein S (named after Seattle), another vitamin K-dependent protein synthesized in endothelial cells, megakaryocytes and hepatocytes, facilitates the action of protein C.
Tertiary Haemostasis (Fibrinolysis)
Tertiary haemostasis is the formation of plasmin from plasminogen via tissue-type plasminogen activators (t-PAs); other plasminogen activators include urokinase, factor XII and kallikrein. Plasmin lyses both fibrinogen and fibrin, releasing fibrin(ogen) degradation products (FDPs). Activation of fibrinolysis is triggered by fibrin and t-PA, a process regulated by haemostasis inhibitors.
Haemostasis inhibitors (inhibitors of fibrinolysis) include TAFI, alpha2-antiplasmin, histidine-rich glycoprotein and plasminogen activator inhibitors. Amplification of the coagulation cascade from thrombin-mediated activation of factor XI leads to large amounts of thrombin and subsequent activation of TAFI, which prevents the binding of plasminogen to fibrin, thus inhibiting its conversion to plasmin. Alpha2-antiplasmin binds free plasmin and causes its removal by the monocyte–macrophage system, preventing widespread fibrin-olysis. Plasminogen activator inhibitors (PAI-1 and PAI-2) are released by endothelial cells and limit plasmin generation by binding to t-PA.
Drugs that inhibit fibrinolysis (antifibrinolytics) include epsilon aminocaproic acid and tranexamic acid.
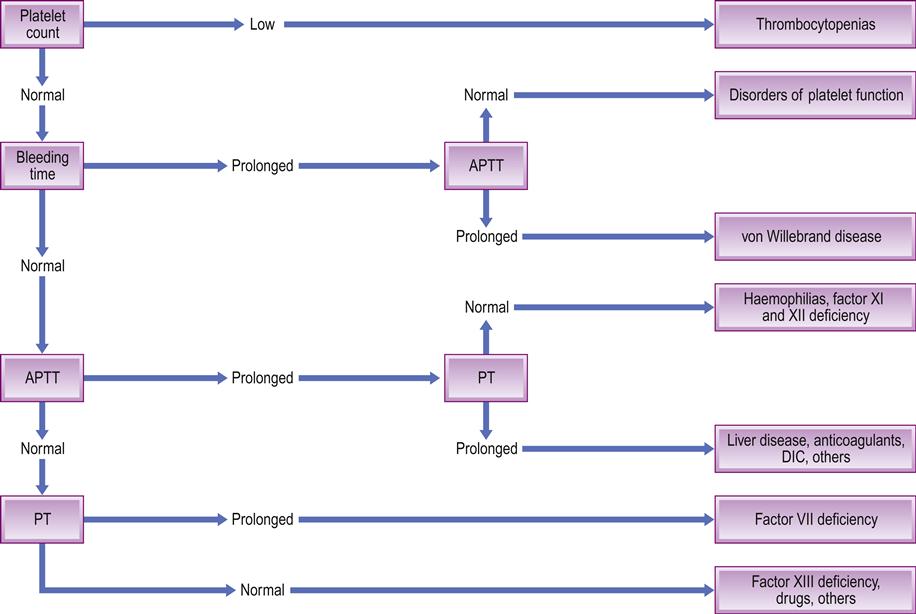
Haemostasis Screening Tests
Screening tests are done to show if there is a bleeding disorder, but further tests needed to define the disorder (Appendix 8.1) must follow consultation with the haematologist to ensure that they are appropriate. All that is needed on the request form is to state: ‘History of prolonged bleeding’ and to ask ‘Would you please investigate haemostatic function?’, giving as much clinical detail as possible. The blood sample should be adequately labelled and sent immediately for testing, together with relevant clinical information. Tests may need to be repeated before an accurate diagnosis can be made because levels of factors vary over time as a result of stress, pregnancy and infections.
Essential tests usually include:
■ activated partial thromboplastin time (APTT). The APTT measures the intrinsic pathway and factors V, VIII, IX, X and XI (Table 8.2). Normal APTT values range between 25±10 seconds. The APTT was sometimes known as PTT or kaolin cephalin PTT. It is prolonged in heparin treatment, liver disease, haemophilias, disseminated intravascular coagulation (DIC), massive transfusion and some autoimmune disorders
■ prothrombin time (PT) and international normalized ratio (INR). The activity of the extrinsic coagulation pathway (factors II, V, VII and X) is measured by the PT, normal values ranging between 12 and 15 seconds. The PT will vary with the type of thromboplastin (e.g. rabbit, human, bovine, etc.) used in the assay, so therefore the INR is used. The INR is the PT ratio of a test sample compared to a normal PT corrected for the sensitivity of the thromboplastin used. Normal INR values are about 1.0. An INR above 1 indicates that clotting will take longer than normal. An INR of 2–3 is the usual therapeutic range for patients on anticoagulants used to treat deep vein thrombosis, and an INR of up to 3.5 is required for patients with prosthetic heart valves. The INR (and PT) is prolonged in warfarin treatment, liver disease, vitamin K deficiency, and DIC (Table 8.3). The INR may be quite stable or erratic in an individual but, in any event, is best assayed less than 24 hours preoperatively, though less than 72 hours may suffice. A normal PT does not necessarily exclude a significant underlying coagulopathy; for example, the PT is normal in severe haemophilias A and B and in factor XI deficiency
■ thrombin time (TT) (citrated sample)
■ platelet count. In the tourniquet test (Hess test), the appearance of more than a few petechiae on the forearm when a sphygmomanometer cuff is inflated suggests a platelet or vascular defect, but the clinical test is neither sensitive nor specific. The ‘bleeding time’ is also rarely used, as it is not completely reliable. Thus to exclude platelet defects, a platelet count is essential and platelet function assays may be indicated. A normal platelet count should be 100 000–400 000 cells/mm3 (Fig. 8.4); a count of less than 100 000 cells/mm3 (thrombocytopenia) can be associated with major postoperative bleeding
■ serum for blood grouping and cross-matching. This should be done if an operation is planned.
Table 8.2
Interpretation of activated partial thromboplastin time (APTT) results
| APTT | Interpretation |
| Isolated prolonged | Acquired clotting factor inhibitors – usually directed against factor VIII (e.g. acquired haemophilia A) |
| Congenital deficiencies of factor XII, XI, IX or VIII (in general, the factor has to be lower than 20–40% of normal before APTT is prolonged) | |
| Lupus anticoagulant (LA) – targets phospholipid | |
| Prolonged+prolonged PT | Combined clotting factors deficiency |
| Disseminated intravascular coagulation (DIC) | |
| Liver disease | |
| Massive blood transfusion – dilutional coagulopathy | |
| Thrombin inhibitors (hirudin, argatroban and dabigatran) | |
| Thrombolytic therapy | |
| Vitamin K deficiency | |
| Increased±prolonged PT | Unfractionated heparin [UFH] significantly prolongs APTT but PT usually shows little prolongation Antiphospholipid antibodies |
| Acquired clotting factor inhibitors, e.g. factors V, X | |
| Prolonged±prolonged PT | Warfarin overdose |
| Short | An acute phase response with high factor VIII |
Table 8.3
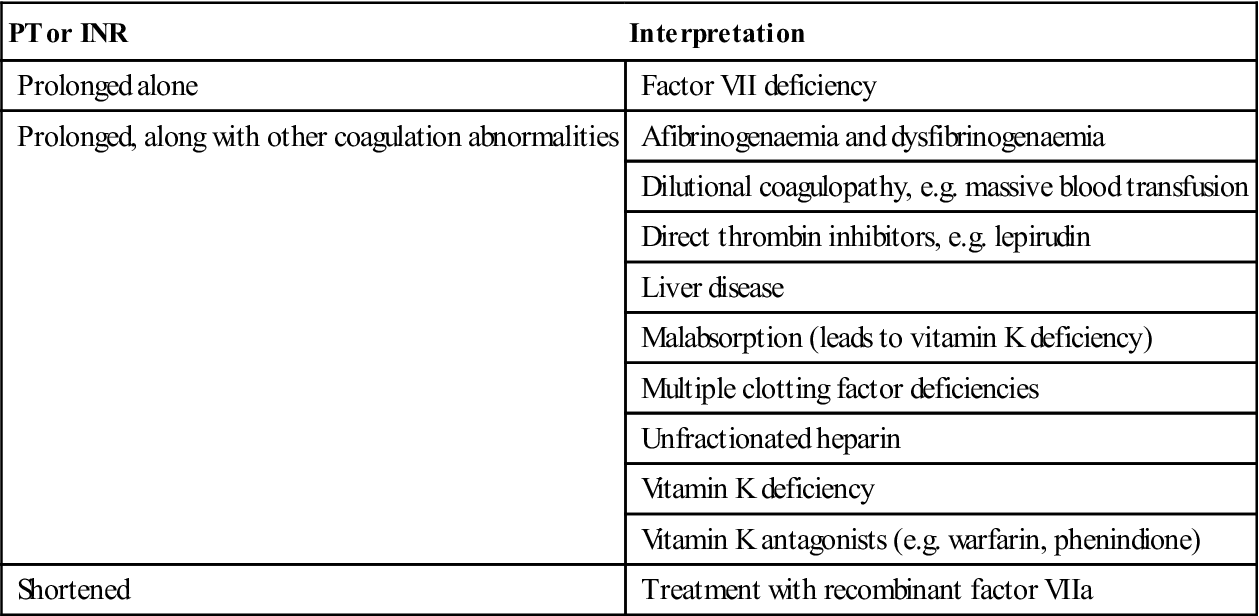
Interpretation of international normalized ratio (INR) and prothrombin time (PT) results
< ?comst?>
| PT or INR | Interpretation |
| Prolonged alone | Factor VII deficiency |
| Prolonged, along with other coagulation abnormalities | Afibrinogenaemia and dysfibrinogenaemia |
| Dilutional coagulopathy, e.g. massive blood transfusion | |
| Direct thrombin inhibitors, e.g. lepirudin | |
| Liver disease | |
| Malabsorption (leads to vitamin K deficiency) | |
| Multiple clotting factor deficiencies | |
| Unfractionated heparin | |
| Vitamin K deficiency | |
| Vitamin K antagonists (e.g. warfarin, phenindione) | |
| Shortened | Treatment with recombinant factor VIIa |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
DIC=disseminated intravascular coagulation.
Once the haemostasis screening tests have indicated that there is a bleeding disorder, other, more specific tests are required, such as:
■ factor VIII clotting activity – to measure factor VIII amount
■ vWF antigen – to measure vWF amount
■ ristocetin cofactor activity – to measure vWF function
A practical guide to laboratory haemostasis is provided at: http://www.practical-haemostasis.com/Screening%20Tests/tt.html (accessed 30 September 2013).
Bleeding (Haemostatic) Disorders
General aspects
Bleeding disorders manifest with common bleeding features, including epistaxis and gingival bleeding, and may occur after wounds and surgery; these are often the first indications of a haemostatic defect. Defects in haemostasis can comprise abnormalities in platelet activation and function, contact activation or with clotting proteins, or may signal excess antithrombin function. The more common causes of a bleeding tendency include:
Clinical features
Examination may reveal signs of purpura in the skin or mucosae (Fig. 8.5). Signs of underlying disease – such as anaemia and lymphadenopathy in leukaemia, for example – must also be sought. Joint deformities from haemarthroses, characteristic of haemophilia, should also be looked for but are infrequently seen now (Fig. 8.6).
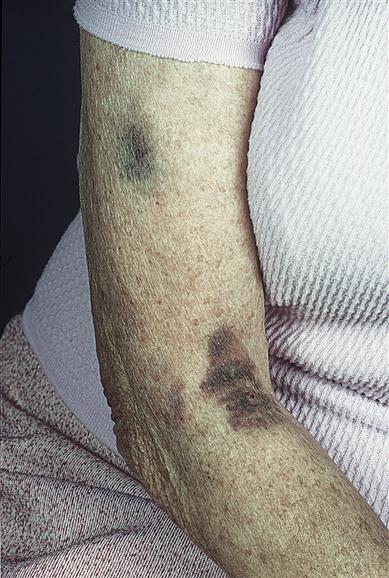
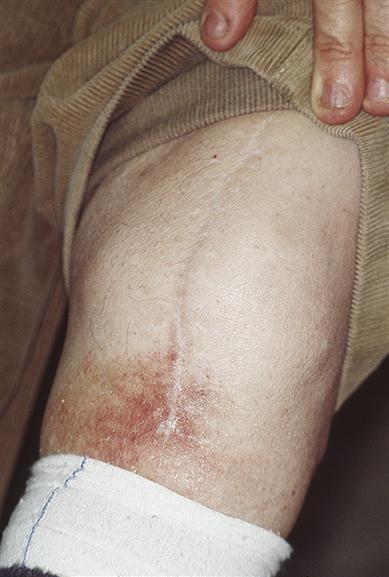
His five brothers all succumbed to haemophilia at a time when factor VIII was unknown.
Alternatively, purpura may be localized to the mouth (sometimes grandiloquently termed ‘angina bullosa haemorrhagica’) and not associated with any abnormal bleeding tendencies. People with senile purpura (an age-related condition) bruise easily with minimal trauma, often without any other apparent underlying cause, and generally on the forearms and tops of the hands. The resulting dark purplish-red splotches fade gradually, often leaving a yellow/brown stain, which may disappear completely or remain. Such purpura can be caused by excessive sun exposure; drugs, such as aspirin and steroids (steroid purpura); diabetes; vascular diseases; thrombocytopenia; scurvy; and connective tissue diseases. There is no treatment available.
General management
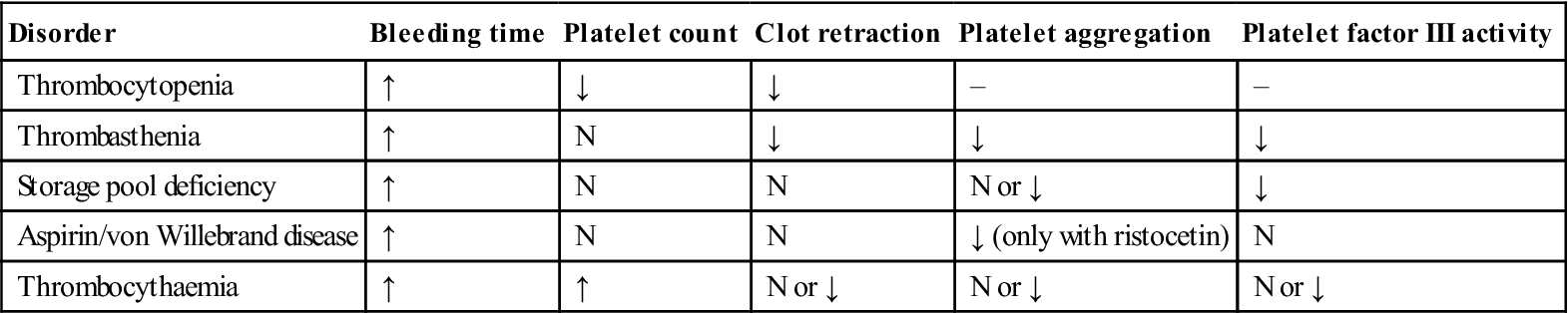
Patients may already be aware of having a bleeding disorder and may carry an appropriate warning card, bracelet or wristband. Otherwise, an adequate history is the single most important part of evaluation; physical examination is necessary and laboratory tests are needed to confirm the diagnosis. Table 8.4 shows comparative features of coagulation defects and platelet defects, and Table 8.5 lists typical laboratory findings in platelet disorders.
Table 8.4
Comparative features of bleeding disorders
| Platelet defectsa | Coagulation defects | |
| Gender affected | Females mainly | Males |
| Family history | Rarely | Usually positive |
| Nature of bleeding after trauma | Immediate | Delayed |
| Effect when locally applied pressure removed | May stop bleeding | Bleeding recurs |
| Spontaneous bleeding into skin or mucosa, or from mucosa or gingivae | Common | Uncommon |
| Bleeding from minor superficial injuries (e.g. needle prick) | Common | Uncommon |
| Deep haemorrhages or haemarthroses | Rare | Common |
| Bleeding time | Prolonged | Normal |
| Tourniquet (Hess) test | Positive | Negative |
| Platelet count | Often low | Normal |
| Clotting function | Normal | Abnormal |
Table 8.5
Typical laboratory findings in platelet disorders
< ?comst?>
| Disorder | Bleeding time | Platelet count | Clot retraction | Platelet aggregation | Platelet factor III activity |
| Thrombocytopenia | ↑ | ↓ | ↓ | – | – |
| Thrombasthenia | ↑ | N | ↓ | ↓ | ↓ |
| Storage pool deficiency | ↑ | N | N | N or ↓ | ↓ |
| Aspirin/von Willebrand disease | ↑ | N | N | ↓ (only with ristocetin) | N |
| Thrombocythaemia | ↑ | ↑ | N or ↓ | N or ↓ | N or ↓ |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
A history with any suggestion of a haemorrhagic tendency must be taken seriously. Nevertheless, patients can be remarkably capricious as to the information they provide and, in any case, cannot always be expected to know when bleeding can legitimately be regarded as ‘abnormal’. Previous dental extractions provide a useful guide, but prolonged bleeding (up to 24–48 hours) as an isolated episode is usually the result of local factors, especially excessive trauma. It should be stressed again that a history of previous haemorrhagic episodes is the most important feature since screening tests of haemostasis do not always detect mild defects.
Special emphasis must be placed on the following, which are suggestive of a bleeding disorder:
■ Deep haemorrhage into muscles, joints or skin – suggests a clotting defect
■ Bleeding from and into mucosae – suggests purpura (Fig. 8.7).
Females often ‘bruise easily’ but any such bruises are usually insignificant and small. Excessive menstrual bleeding is also rarely due to a bleeding disorder.
Most significant congenital bleeding disorders become apparent in childhood and patients may carry a warning card. However, people with mild bleeding tendencies can escape recognition until adult life if they manage to avoid injury or surgery, but can then present with oozing from an extraction socket for 2–3 weeks despite local haemostatic measures such as suturing. Patients who have had tonsillectomy or dental extractions without trouble, or previous dental bleeding that was controlled by local measures, are unlikely to have a serious congenital haemorrhagic disease. On the other hand, admission to hospital and blood transfusion or comparable measures have obvious implications. Haemorrhagic disease in a blood relative is also strongly suggestive of a clotting defect. Many drugs, such as anticoagulants (Fig. 8.8), NSAIDs and antiplatelet drugs, may cause bleeding tendencies, as may hepatic, renal, human immunodeficiency (HIV) and other disease. Some herbal products may also impair platelet aggregation and prolong bleeding (Ch. 26).

Precise characterization of a congenital clotting defect depends on assay of the individual factors, and a wide range of other investigations may be indicated according to the type of case. For example, the assays usually used in the diagnosis of haemophilia A are the APTT and the factor VIII coagulant (FVIIIC) activity. The bleeding time and whole-blood clotting time are uninformative and obsolete.
It is important also to investigate patients with a suspected bleeding tendency for anaemia. This is because:
■ anaemia is an expected consequence of repeated haemorrhages
■ anaemia may be an essential concomitant of the haemorrhagic tendency – as in leukaemia.
Patients may also need to be screened for liver disease and for blood-borne infections, particularly HIV and hepatitis viruses.
Individuals with bleeding disorders may need to be treated with replacement of the missing factor. Earlier, use of blood or blood fractions sometimes resulted in the transmission of blood-borne viruses and other infections. In many countries, blood is now collected from donors under careful precautions, and screened to exclude antibodies to infections such as HIV, hepatitis B, hepatitis C, syphilis or cytomegalovirus. In developed countries, all cellular blood products are leukodepleted to limit the risk of transmission of prions and some other infections. Leukocytes in erythrocyte and platelet concentrates are now considered as a contaminant since they can cause many other adverse effects, such as the transmission of other cell-associated infectious agents (e.g. herpesviruses and Toxoplasma), febrile non-haemolytic reactions, graft-versus-host disease and immunosuppression. Recombinant factors are thus preferred for factor replacement.
Dental aspects
Prolonged post-operative bleeding is defined if any of the following appertains:
■ Bleeding continues for 12 hours.
■ Bleeding causes the patient to return for attention.
All dental professionals encounter patients who experience prolonged bleeding following operative procedures, often a dental extraction. Saliva contains fibrinolytic agents that may aggravate the situation. In most cases, the cause is local, and bleeding can be managed using simple local haemostatic measures such as applying pressure to the wound with sterile pads (moistened with water, normal saline or 5% tranexamic acid solution), using absorbable oxidized cellulose sponges, and suturing, as well as giving postoperative instructions verbally and in writing.
A bleeding tendency can be encountered in patients with congenital haemostatic defects such as haemophilias or some rare platelet defects; or acquired disorders such as may arise in liver disease, renal disease, bone marrow disease, immune disorders or with some drugs. In vascular or platelet disorders, postoperative problems following an extraction usually present as prolonged bleeding immediately after the event; in coagulation defects, the bleeding typically begins after some delay. Patients with a bleeding tendency may require prophylactic measures preoperatively, special precautions perioperatively and careful management postoperatively. Patients with hereditary bleeding disorders are often registered with a haemophilia reference centre. Care is as follows:
Some known haemophiliacs have neglected oral health because of barriers to accessing care, including travel to specialist centres, waiting times, increased treatment needs and cost, or fear of bleeding from dental procedures. They may require more complex treatment and management by the time that they present.
Surgery should ideally be planned and done where appropriate after suitable measures to correct the bleeding tendency, such as blood clotting factor or platelet replacement or, where drugs are responsible, by modifying drug doses provided that the risk of subsequent thromboembolism is not a contra-indication:
Vascular Disorders
Vascular purpura rarely causes serious bleeding; any bleeding into mucous membranes or skin starts immediately after trauma but stops within 24–48 hours. Vascular disorders are a rare cause of bleeding problems; they include Marfan, Ehlers–Danlos (Ch. 16) and Osler–Rendu–Weber syndromes.
Osler–Weber–Rendu Syndrome (Hereditary Haemorrhagic Telangiectasia [HHT/HHT1])
General aspects
Hereditary haemorrhagic telangiectasia (HHT) is an autosomal dominant condition with a high penetrance, as 97% of people affected exhibit symptoms. There is vascular dysplasia leading to telangiectasia and arteriovenous malformations in the skin, mucosa and viscera. There may be associated immunoglobulin A (IgA) deficiency or, rarely, vWD (see below).
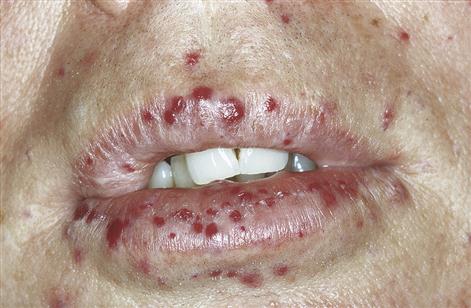
Clinical features
Interestingly, HHT usually presents in adolescence. Telangiectasia on the skin or any part of the oral, nasal, conjunctival, respiratory, gastro-intestinal or urogenital mucosa, or brain and liver (Fig. 8.9), leads to bleeding. Most people (62%) are diagnosed by the age of 16, with over 90% of cases presenting with recurrent epistaxis, not present until adult life. Other associated features include:
General management
Investigations include capillary microscopy, computed tomography (CT), magnetic resonance imaging (MRI) and angiography. Treatment is by cryosurgery or argon laser treatment, if the telangiectases have bled significantly or are cosmetically unacceptable.
Dental aspects
Telangiectases may be seen in any part of the mouth and may be conspicuous on the lips and tongue. Bleeding from oral surgery is unlikely to be troublesome. Rarely, brain abscesses or other infections have followed oral procedures that cause bacteraemias but there is no consensus as to the need for antibacterial prophylaxis.
Regional local anaesthesia (LA) should be avoided because of the risk of deep bleeding. Conscious sedation (CS) can be given if required. In general anaesthesia (GA), nasal intubation is best avoided and close postoperative observation is advisable.
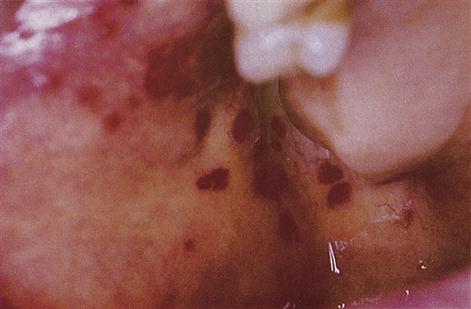
Localized Oral Purpura (‘Angina Bullosa Haemorrhagica’)
Blood blisters are occasionally seen, typically in older persons, in the absence of obvious trauma, generalized purpura, any other bleeding tendency or evidence of any systemic disease. These blood blisters are often in the palate and may sometimes be a centimetre or more in diameter; after rupture, they may leave a sore area for a few days. When a large blood blister of this type is in the pharynx, it can cause an alarming choking sensation and was therefore originally termed ‘angina bullosa haemorrhagica’. However, almost any site in the mouth can be affected.
Before the diagnosis of localized oral purpura can be made confidently and the patient reassured, other causes of blood blisters in the mouth must be excluded. These include: platelet disorders, amyloidosis (causing factor X deficiency), leukaemia, infectious mononucleosis, HIV or rubella; trauma; corticosteroid inhalers; or bleeding into subepithelial blisters, such as in mucous membrane pemphigoid (Ch. 11).
Platelet Disorders
Platelets originate from bone marrow megakaryocytes and have a life span of about 1 week, before they are destroyed in the spleen. Thrombopoietin, the megakaryocyte-stimulating hormone is produced in the liver and kidneys, so liver, kidney or bone marrow disease can each cause thrombocytopenia. Platelet numbers or function, if impaired, can cause a bleeding tendency, purpura and abnormal laboratory investigation results (Tables 8.5 and 8.6). Drugs, bone marrow invasion, autoimmune diseases and other disorders can reduce platelet numbers (if the platelet count falls below 100×109/L, a patient has thrombocytopenia). Thrombocytopenia may arise from (Fig. 8.10):
■ sequestration of platelets. An enlarged spleen (splenomegaly) is usually responsible for platelet sequestration which may be, therefore, caused by:
portal hypertension, e.g. cirrhosis
haematological disorders, e.g. myeloproliferative disorders
infections, e.g. malaria, leishmaniasis, Epstein–Barr virus
storage disorders, e.g. Gaucher disease, Niemann–Pick disease
inflammatory disorders, e.g. systemic lupus erythematosus (SLE), Felty syndrome.
Table 8.6
< ?comst?>
| Defect | Manifestations | Management | |
| Thrombasthenia (Glanzmann syndrome) | Defective platelet aggregation due to defective membrane protein | Severe bleeding tendency | Platelet infusions needed preoperatively |
| Bernard–Soulier syndrome | Giant platelets with defect in platelet glycoprotein which acts as receptor for von Willebrand factor | Heritable disorder resembling von Willebrand disease but considerably more rare | Platelet infusions needed preoperatively |
| Storage pool deficiency | Platelets lack capacity to store serotonin and adenine nucleotides, and consequently fail to aggregate | Usually congenital with autosomal inheritance. If associated with albinism, is termed Hermansky–Pudlak syndrome | Platelet infusion or cryoprecipitate corrects the bleeding tendency |
| HIV infection | Purpura is a relatively common feature of HIV infection. May be autoimmune but there appears also to be a platelet defect resulting from the infection; drugs such as zidovudine or some protease inhibitors and bone marrow disease may also contribute | Oral purpura in HIV infection may closely mimic oral lesions of Kaposi sarcoma (Ch. 20) | See Ch. 20 |
| Chronic renal failure | Ch. 12 | ||
| Drugs: heparin, dextrans | See later in this chapter | ||
| Dysproteinaemias | See later in this chapter | ||
| Myelodysplastic syndrome | See later in this chapter | ||
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Inherited platelet abnormalities tend to affect platelet function rather than number. Abnormal platelet function is seen in Glanzmann disease and dysproteinaemias. Platelet function can also be impaired due to an increase in numbers (thrombocytosis). Abnormal platelet distribution is seen in splenomegaly or transfusion of stored blood.
Idiopathic Thrombocytopenic Purpura (ITP)
This involves autoantibody-mediated platelet destruction, the most common antibody being directed against the platelet GPIIb–IIIa receptor. Features include petechiae, ecchymoses and postoperative haemorrhage. Full blood picture and platelet counts are indicated. Corticosteroids or other immunosuppressives are the main treatments. Splenectomy or splenic irradiation, followed by thrombopoietin therapy is sometimes needed. Eltrombopag and romiplostin are thrombopoietin receptor agonists which stimulate platelet production and used to treat ITP. Dental extractions can be covered by these agents.
Haemolytic Uraemic Syndrome (HUS) and Thrombotic Thrombocytopenic Purpura (TTP)
These conditions are caused by small-vessel wall damage, leading to fibrin and platelet deposition and haemolysis. HUS predominantly affects the kidneys and TTP the brain. Escherichia coli O157 gastroenteritis may cause HUS in children, but in adults more commonly causes TTP.
Glanzmann Disease (Glanzmann Thrombasthenia)
Glanzmann disease is a rare autosomal recessive disorder caused by either deficient or abnormal glycoprotein platelet receptors. Defects in the GP IIb/IIIa complex lead to defective platelet aggregation and subsequent bleeding, usually presenting in infancy or early childhood, with bruising or petechiae following minimal or unrecognized trauma, or epistaxis. The platelet count is normal but platelet aggregation defective.
Management includes avoiding trauma and avoiding analgesics such as aspirin. Minor bleeding intraorally can be managed with tranexamic acid. Major bleeding may require platelet transfusion. Recombinant human-activated factor VII (rFVII) has been used in patients with antibodies to GP IIb/IIIa and/or human leukocyte antigen (HLA) that render transfusions ineffective.
Bernard–Soulier Syndrome
Bernard–Soulier syndrome is a rare congenital disorder with large platelets that lack platelet membrane glycoproteins – resulting in defective platelet adhesion. Clinical features resemble those of Glanzmann disease. A full blood count reveals giant platelets. Platelets fail to aggregate in response to vWF.
Management is as for Glanzmann disease.
Renal Failure
High serum urea levels directly inhibit platelet function and there may also be thrombocytopenia. Desmopressin (deamino-8-D-arginine vasopressin; DDAVP) may be used to boost vWF levels in order to treat mild bleeding episodes. In severe bleeding, cryoprecipitate or platelet transfusion may be required.
Antiplatelet Drugs
Platelets may pathologically aggregate intravascularly as arterial thrombi in response to haemorrhage into atherosclerotic plaques, and this is potentially lethal. Antiplatelet drugs prevent and/or reverse platelet aggregation in arterial thromboses, and help prevent clotting in patients who have had a myocardial infarct, unstable angina, ischaemic strokes, transient ischaemic attacks (TIAs or ‘little strokes’) and other forms of cardiovascular disease. In practice, a favourable balance between the beneficial and harmful effects of antiplatelet therapy is achieved by treating patients whose thrombotic risk outweighs their risk of bleeding complications.
Aspirin
Aspirin is an irreversible inhibitor of platelet cyclo-oxygenase (COX) enzymes, which catalyse the production of thromboxane A2 (an important platelet aggregator) from arachidonic acid. Low-dose aspirin is the usual first-line treatment for thrombosis and myocardial infarction. The most common adverse effect of aspirin is gastrointestinal bleeding, which can be minimized by using the lowest effective dose (often 75 mg daily), advising patients to take aspirin with food, and, where indicated, co-prescribing a proton pump inhibitor (PPI).
GP IIb/IIIa inhibitors
These drugs prevent cross-linking of platelets. Abciximab is a chimeric human–murine GP IIb/IIIa antibody; the peptide derivatives, eptifibatide and tirofiban, are more selective towards the GP IIb/IIIa receptor and have a shorter effect than abciximab. These agents are used parenterally in acute coronary syndromes, but only by specialists. Serious adverse effects of GP IIb/IIIa antagonists include major bleeding, intracerebral haemorrhage and thrombocytopenia.
Phosphodiesterase inhibitors
Phosphodiesterase inhibitors include dipyridamole – a vasodilator and antiplatelet agent that inhibits adenosine uptake and cyclic guanosine monophosphate (cGMP) phosphodiesterase activity, thus decreasing platelet aggregability. Dipyridamole is used with aspirin or warfarin in the prophylaxis of thromboembolic disorders.
Thienopyridines
Thienopyridines act via inhibition of platelet activation by the ADP-dependent pathway without a direct effect on prostaglandin metabolism. Ticlopidine is the oldest thienopyridine available and is approved for secondary prevention of thrombotic strokes in aspirin-intolerant patients, and in combination with aspirin for prevention of coronary stent thrombosis. Serious adverse effects are mainly haematological (neutropenia, thrombocytopenia and TTP). Clopidogrel has a better safety profile than ticlopidine and is often reserved for those with true aspirin hypersensitivity or intolerance, and for prevention of atherosclerotic events following recent myocardial infarction, stroke or established peripheral arterial disease. It is also approved for use in acute coronary syndromes that are treated with either percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG). Dual therapy with aspirin and clopidogrel increases the risk of gastrointestinal bleeding and is thus usually restricted to patients following PCI or acute coronary syndromes. Prasugrel is a more rapid-acting and potent platelet aggregation inhibitor and may be used together with aspirin in the setting of high-risk or urgent PCI.
Dental aspects of platelet disorders
The main danger of platelet disorders is haemorrhage but this is rarely as severe as in clotting disorders. Regional LA block injections can be given if the platelet levels are above 30×109/L. Haemostasis after dentoalveolar surgery is usually adequate if platelet levels are above 50×109/L. Major surgery requires platelet levels above 75×109/L. CS can be given but if it is delivered by the intravenous route, care must be taken not to damage the vein. GA can be given in hospital but expert intubation is needed to avoid the risk of submucous bleeding into the airway.
Platelets can be replaced or supplemented by platelet transfusions (1 unit of platelets should raise the count by around 10×109 platelets/L), but sequestration of platelets is very rapid. Platelet transfusions are therefore best used for controlling already established thrombocytopenic bleeding. When given prophylactically, platelets should be given in the following way: half just before surgery to control capillary bleeding, and half at the end of the operation to facilitate placement of adequate sutures. Platelets should be used within 6–24 hours of collection. Suitable preparations include platelet-rich plasma (PRP), which contains about 90% of the platelets from a unit of fresh blood in about half this volume, and platelet-rich concentrate (PRC), which contains about 50% of the platelets from a unit of fresh whole blood in a volume of only 25 mL. PRC is thus the best source of platelets. Platelet infusions carry the risk of isoimmunization, infection with blood-borne agents and, rarely, graft-versus-host disease. Where there is immune destruction of platelets (e.g. in ITP), platelet infusions are less effective. The need for platelet transfusions can be reduced by local haemostatic measures and use of desmopressin (DDAVP) or tranexamic acid or topical administration of platelet concentrates. Absorbable haemostatic agents, such as oxidized regenerated cellulose (Surgicel), synthetic collagen (Instat) or microcrystalline collagen (Avitene), may be put in the socket to assist clotting.
Splenectomy predisposes to infections, typically with pneumococci, and especially within the first 2 years. Systemic infection post-splenectomy, involving oral streptococci, is rare and antimicrobial prophylaxis is not therefore generally recommended before invasive dental procedures.
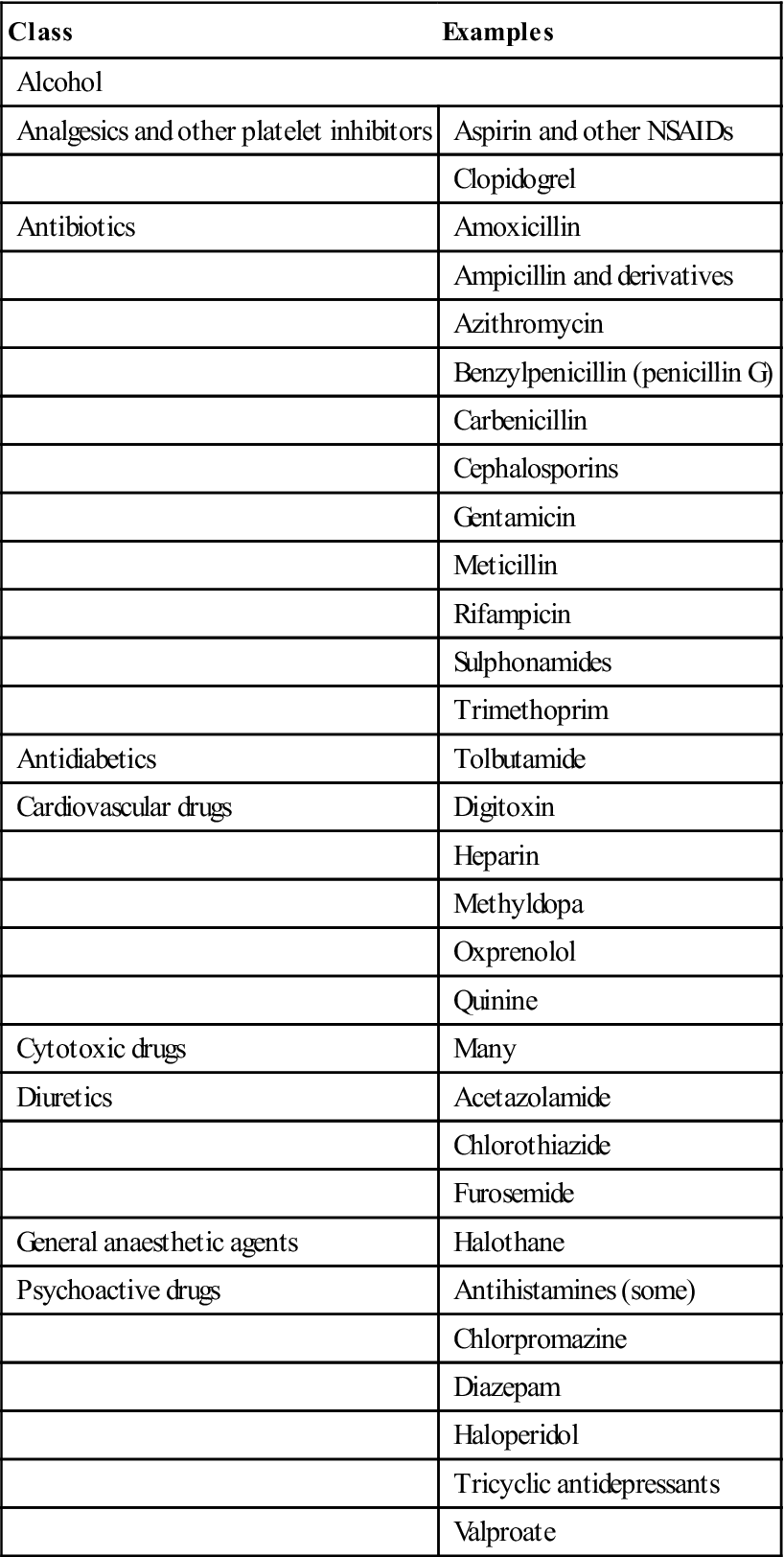
Long-term corticosteroids can cause well-recognized problems (Ch. 6). Perioperative management is summarized in Tables 8.6 and 8.7 and Figure 8.11. Drugs such as aspirin and other NSAIDs that damage platelets should be avoided (Table 8.8). The effects of aspirin are not dose-dependent; even a single tablet can affect platelet COX irreversibly for about a week. Other NSAIDs may have a reversible effect and act for up to 48 hours only. COX-2 inhibitors have no such effect on platelets.
Table 8.7
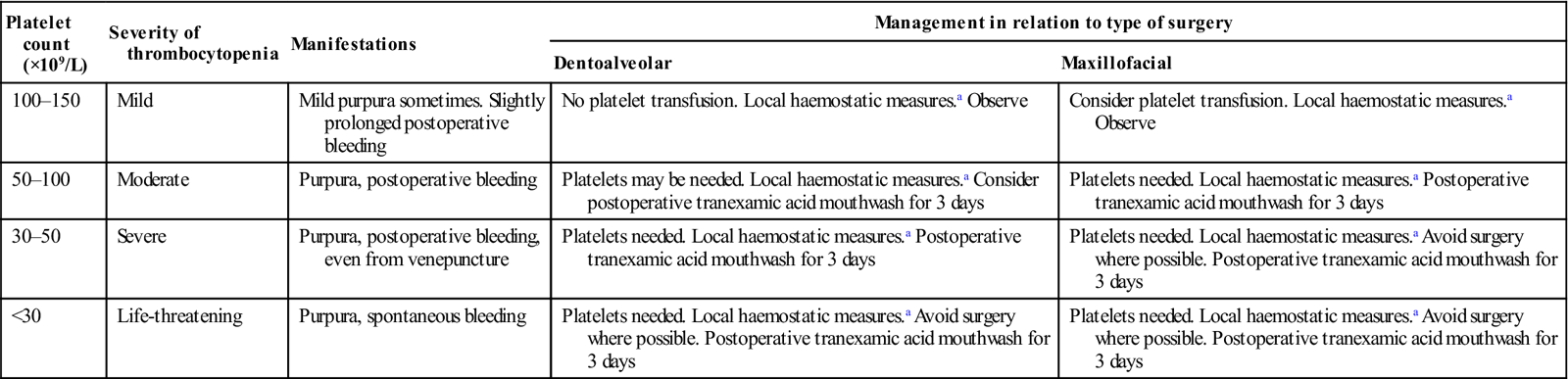
Thrombocytopenia: manifestations and management of oral surgery
< ?comst?>
| Platelet count (×109/L) | Severity of thrombocytopenia | Manifestations | Management in relation to type of surgery | |
| Dentoalveolar | Maxillofacial | |||
| 100–150 | Mild | Mild purpura sometimes. Slightly prolonged postoperative bleeding | No platelet transfusion. Local haemostatic measures.a Observe | Consider platelet transfusion. Local haemostatic measures.a Observe |
| 50–100 | Moderate | Purpura, postoperative bleeding | Platelets may be needed. Local haemostatic measures.a Consider postoperative tranexamic acid mouthwash for 3 days | Platelets needed. Local haemostatic measures.a Postoperative tranexamic acid mouthwash for 3 days |
| 30–50 | Severe | Purpura, postoperative bleeding, even from venepuncture | Platelets needed. Local haemostatic measures.a Postoperative tranexamic acid mouthwash for 3 days | Platelets needed. Local haemostatic measures.a Avoid surgery where possible. Postoperative tranexamic acid mouthwash for 3 days |
| <30 | Life-threatening | Purpura, spontaneous bleeding | Platelets needed. Local haemostatic measures.a Avoid surgery where possible. Postoperative tranexamic acid mouthwash for 3 days | Platelets needed. Local haemostatic measures.a Avoid surgery where possible. Postoperative tranexamic acid mouthwash for 3 days |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Table 8.8
Some drugs that may impair platelets or their function
< ?comst?>
| Class | Examples |
| Alcohol | |
| Analgesics and other platelet inhibitors | Aspirin and other NSAIDs |
| Clopidogrel | |
| Antibiotics | Amoxicillin |
| Ampicillin and derivatives | |
| Azithromycin | |
| Benzylpenicillin (penicillin G) | |
| Carbenicillin | |
| Cephalosporins | |
| Gentamicin | |
| Meticillin | |
| Rifampicin | |
| Sulphonamides | |
| Trimethoprim | |
| Antidiabetics | Tolbutamide |
| Cardiovascular drugs | Digitoxin |
| Heparin | |
| Methyldopa | |
| Oxprenolol | |
| Quinine | |
| Cytotoxic drugs | Many |
| Diuretics | Acetazolamide |
| Chlorothiazide | |
| Furosemide | |
| General anaesthetic agents | Halothane |
| Psychoactive drugs | Antihistamines (some) |
| Chlorpromazine | |
| Diazepam | |
| Haloperidol | |
| Tricyclic antidepressants | |
| Valproate | |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
When prescribed as primary prevention, antiplatelet agents may be safely withdrawn 7 days before surgery, but any decision to stop antiplatelet therapy used for secondary prevention must balance the risk of thrombosis and ischaemia against bleeding. There is no evidence that continuing antiplatelet monotherapy causes major bleeding problems during or after minor surgery such as dental extractions. The risk of bleeding is greater with aspirin–clopidogrel or prasugrel–aspirin combinations, but patients taking these drugs are generally already at a higher risk of thromboembolic events; therefore, advice from a specialist should be sought before withholding treatment, or the patient may need to be referred to a hospital setting for the procedure. There is no suitable test available to assess the increased risk of bleeding in patients taking antiplatelet medications.
Patients who should not have their medications stopped or altered prior to dental surgical procedures in primary care include those taking:
Patients are more at risk of permanent disability or death from thromboembolic episodes if they stop antiplatelet medications prior to a surgical procedure than if they continue them. Although the risk is low, the outcome is serious, so antiplatelet medications should only be discontinued in the perioperative period when the haemorrhagic risk of continuing them is definitely greater than the cardiovascular risk associated with their discontinuation. Bleeding complications, while inconvenient, do not carry the same risks as thromboembolic complications. Patients taking antiplatelet medications will have a prolonged bleeding time but this may not be clinically relevant and postoperative bleeding after dental procedures can usually be controlled using local haemostatic measures.
Consensus, then, is that for dentoalveolar surgical procedures, antiplatelet medications should not be stopped nor doses altered, but that local haemostatic measures should be used to control bleeding. The following patients taking antiplatelet medication, however, should not be treated in primary care without medical advice – or should be referred to a dental hospital or hospital-based dental clinic:
■ Those with liver impairment and/or alcoholism
■ Those with thrombocytopenia, haemophilia or other disorder of haemostasis
■ Those currently receiving a course of cytotoxic medication.
Dentoalveolar surgical procedures likely to be carried out in primary care will be classified as minor, e.g. simple extraction of up to three teeth, gingival surgery, crown and bridge procedures, dental scaling and the surgical removal of teeth. When more than three teeth need to be extracted, multiple visits will be required. Extractions may be planned to remove two or three teeth at a time, by quadrants, or singly at separate visits. Scaling and gingival surgery should initially be restricted to a limited area to assess whether bleeding is problematic. Patients requiring major surgery should usually be treated in a secondary care setting.
Blood Coagulation Disorders
Blood coagulates via a number of liver-synthesized serum proteins (coagulation factors), activated by a cascade of enzymatic reactions. Two pathways of coagulation – the intrinsic and extrinsic – have a common end-point at the formation of thrombin, which then catalyses the formation of fibrin from fibrinogen. Coagulation disorders – typically from drugs (e.g. warfarin), liver failure and renal failure – are the main causes of such a bleeding tendency.
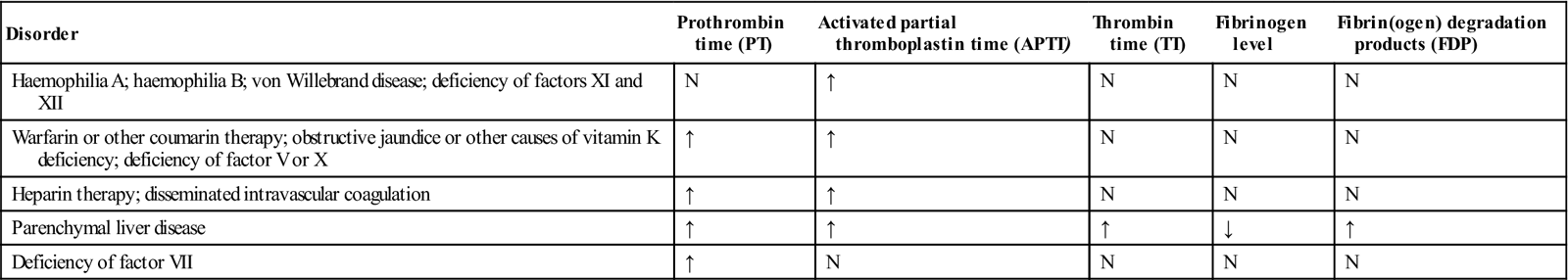
Coagulation disorders are due mainly to genetic defects of clotting factors (vWD but also other haemophilias), to anticoagulant or other drug therapy (Table 8.9), or to a range of diseases, especially those affecting the liver or kidney (Box 8.1). These cause a range of changes in the results of laboratory investigations (Table 8.10).
Table 8.9
Drug effects on bleeding tendency
| Drug | Effect on bleeding |
| Alcohol | Warfarin enhanced by large amounts of alcohol |
| Analgesics | Bleeding enhanced by aspirin effect on platelets |
| Antibacterials | Warfarin enhanced by cephalosporins, erythromycin and metronidazole. Ampicillin and amoxicillin may increase bleeding |
| Antifungals | Warfarin enhanced by azoles, including miconazole topically |
| Anti-inflammatories | Bleeding enhanced by antiplatelet activity of NSAIDs; warfarin may also be enhanced. Corticosteroids may alter warfarin activity |
Table 8.10
Laboratory findings in clotting disorders
< ?comst?>
| Disorder | Prothrombin time (PT) | Activated partial thromboplastin time (APTT) | Thrombin time (TT) | Fibrinogen level | Fibrin(ogen) degradation products (FDP) |
| Haemophilia A; haemophilia B; von Willebrand disease; deficiency of factors XI and XII | N | ↑ | N | N | N |
| Warfarin or other coumarin therapy; obstructive jaundice or other causes of vitamin K deficiency; deficiency of factor V or X | ↑ | ↑ | N | N | N |
| Heparin therapy; disseminated intravascular coagulation | ↑ | ↑ | N | N | N |
| Parenchymal liver disease | ↑ | ↑ | ↑ | ↓ | ↑ |
| Deficiency of factor VII | ↑ | N | N | N | N |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Congenital Coagulation (Clotting) Disorders (Coagulopathies)
These are usually due to a deficiency or lack of a specific blood- clotting factor. The vascular and platelet haemostatic responses usually mask any problem initially, and patients usually complain of a delayed blood oozing from the area, such as a tooth extraction socket.
The most important hereditary bleeding disorder in terms of prevalence and severity is von Willebrand disease, but haemophilia A and B (Christmas disease) are the other most common coagulopathies. Haemophilia A and B are hereditary X-linked recessive disorders characterized by deficiencies in blood clotting factors VIII and IX, respectively. Factor XI deficiency is also important. Less common disorders are summarized in Table 8.11 and Appendix 8.2.
Table 8.11
Blood coagulation factor defects in descending order of frequency
| Factor deficient | Inheritance | Incidence (1 in) |
| VIII | X-linked | 10 000 |
| IX | X-linked | 60 000 |
| VII | Autosomal recessive | 500 000 |
| V | Autosomal recessive | 1 million |
| X | Autosomal recessive | 1 million |
| XI | Autosomal recessive | 1 million |
| XIII | Autosomal recessive | 1 million |
| Fibrinogen | Autosomal recessive | 1 million |
| Prothrombin | Autosomal recessive | 2 million |
Haemophilia A
Haemophilia A affects approximately 1 in 5000 males.
General aspects
Haemophilia A is an X-linked disorder resulting from a deficiency in blood clotting factor VIII, a key component of the coagulation cascade. Haemophilia A thus affects males. Sons of carriers have a 50:50 chance of developing haemophilia, while daughters of carriers have a 50:50 chance of being carriers. All daughters of an affected male are carriers but sons are normal. Carriers rarely have a clinically manifest bleeding tendency. The female carrier will transmit the disorder to half of her sons and the carrier state to half of her daughters. The affected males will not transmit the disorder to their sons because their Y chromosome cannot carry the haemophilic gene.
Haemophilia A is about ten times as common as haemophilia B, except in some Asians, in whom frequencies are almost equal. Haemophilia A arises from a variety of mutations; some 150 different point mutations have been characterized but a family history can be obtained in only about 65% of cases.
Clinical features
Haemophilia is characterized by excessive bleeding, particularly after trauma and sometimes spontaneously. Haemorrhage appears to stop immediately after the injury (due to normal vascular and platelet haemostatic responses) but intractable oozing with rapid blood loss soon follows. Haemorrhage is dangerous either because of acute blood loss or due to bleeding into tissues, particularly the brain, larynx, pharynx, joints and muscles (Fig. 8.12). Abdominal haemorrhage may simulate an ‘acute abdomen’. Haemarthroses can cause joint damage and cripple the patient.
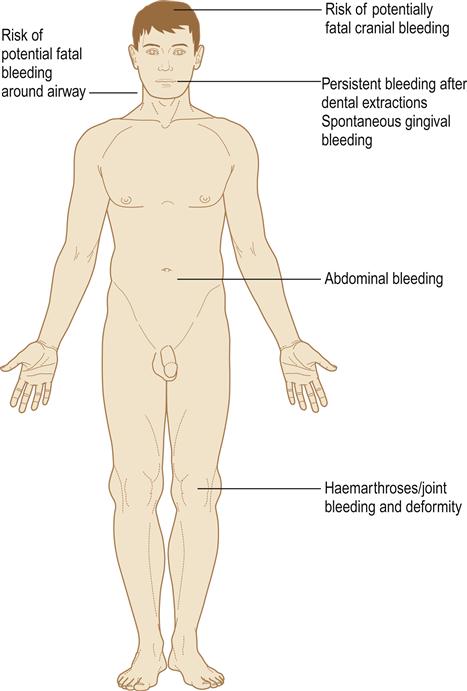
Dental extractions lead to prolonged bleeding and, in the past, have been fatal. Abnormal bleeding after extractions has sometimes led to recognition of haemophilia. The severity of bleeding in haemophilia A correlates with the level of factor VIII:coagulant (VIII:C) activity and degree of trauma (Table 8.12). Normal plasma contains 1 unit of factor VIII per millilitre, a level defined as 100%.
Table 8.12
| % factor VIII | Clinical features | |
| Severe | <1 | Spontaneous bleeding, typically from childhood, with bleeding into muscles or joints (haemarthroses), easy bruising and prolonged bleeding from minor injuries |
| Moderate | 1–5 | Comparatively minor trauma may still lead to significant blood loss |
| Mild | >5–25 | Comparatively minor trauma may still lead to significant blood loss. Bleeding after dental extractions is sometimes the first or only sign of mild disease |
| Very mild | >25 | Patient can generally lead a normal life and may remain undiagnosed but there can be prolonged bleeding after trauma or surgery. Some may not bleed excessively even after a simple dental extraction, so that the absence of post-extraction haemorrhage cannot reliably be used to exclude haemophilia. Most will, however, bleed excessively after more traumatic surgery, such as tonsillectomy |
Haemophilia should be suspected in males presenting with a history of:
Unlike platelet disorders, which present as bleeding into and from mucous membranes, haemophilia presents mainly as bleeding into muscle or joints (haemarthrosis). More seriously, a cerebral bleed can be a fatal complication. Bleeding around the larynx or pharynx after inferior alveolar nerve block administration or floor-of-mouth infiltration can obstruct the airway. While the history of bleeding is usually life-long, some children with severe haemophilia may not have bleeding symptoms until later, when they begin walking or running. Patients with mild haemophilia may not bleed excessively until they experience trauma or surgery. The severity of bleeding in haemophilia is generally correlated with the clotting factor level:
General management
Diagnosis
The diagnosis of haemophilias is based upon the clinical presentation, a positive family history, coagulation studies and clotting factor assays. Typical findings are shown in Box 8.2. In haemophilia, the various factors are:
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


