8 Elastomeric Ligatures and Chains
Bright-field double-transmittance polarized-light images of a chain that had been elongated 50% for 24 hours. (Original magnification × 12.5)
Fluoride-Releasing Elastomerics
In vitro Studies of Force Degradation
Introduction
Elastomeric products are used in orthodontics as ligatures and as continuous modules (chains) for the engagement and retraction of teeth. (The use of the popular elastomeric alginate impression materials in orthodontics will be discussed in Chapter 12.) The elastomeric modules were first introduced to orthodontics nearly three decades ago and have gained almost universal acceptance by the profession. They have replaced the elastic appliances used for the retraction of teeth that were fabricated from latex rubber and required daily replacement by the patient. Moreover, orthodontists prefer use of the elastomeric ligatures rather than stainless steel ligature wires.
Despite this popularity, there has been some concern about the force degradation exhibited by the elastomeric chains, which will be discussed later in this chapter. Because of this concern, over the past decade there has been increasing interest in self-ligating brackets to minimize the inventory required for orthodontic practice and to increase the reliability of the archwire-bracket system. Efforts have also been directed to minimization of the plaque-retaining capacity of the elastomeric chains. Fluoride-releasing elastomeric ligatures have been introduced to minimize the risk for demineralization at the enamel margins. Alternatively, novel retracting assemblies have been developed that instead use rare-earth magnets or NiTi coil springs to provide efficient tooth movement with minimum patient monitoring and significantly longer intervals between treatment, compared to those required with the use of conventional elastomeric chains.
Composition and Structure
The elastomeric ligatures and chains are polyurethanes, which are thermosetting polymers possessing a — (NH) — (C = O) — O — structural unit and formed by step-reaction (condensation) polymerization (Chapter I). Manufacture of the polyurethane elastomers involves several stages. A basic intermediate is first prepared in the form of a low-molecular-weight polymer, usually a polyester or polyether. This intermediate is then reacted with an aromatic diisocya-nate to yield a prepolymer. This elastomer is vulcanized through isocyanate groups by reaction with alcohols or glycols. Detailed information about the synthesis, structure and properties of polyurethanes used for biomedical applications can be found in the book by Lambda et al.
Polymers possessing rubber-like elasticity (Chapter I) have long-chain, lightly cross-linked structures. The restoring force leading to this type of elastic behavior is the result of a decrease in entropy associated with the distortion of a macromolecular chain from its most probable conformation. However, the local mobility of the chain segments must be restricted in order for the polymer to return to its original shape, since irreversible movement of chains past each another will cause permanent deformation of the material. The cross-links between chains must be relatively few to facilitate large extensions with no rupture of primary bonds. The glass transition temperatures (Tg) of industrial and biomedical polyurethanes range from about −50 °C to −80 °C.
Indirect measurement of the structure and mechanical properties of these polymers provides insight into the loss of force. The glass transition temperature, previously described in Chapter 1, is considered a fundamental property of a polymer and can be measured using thermal analysis techniques. Chapter 3 discussed the use of differential scanning calorimetry (DSC) to determine Tg for a polymer. Knowledge of the value of Tg often leads to plausible inferences about the polymer structure and mechanical behavior. The glass transition occurs over a range of temperatures at which the polymer transforms from a rigid or glassy state to a rubbery state. For convenience, the glass transition temperature is generally defined as the midpoint of this temperature range, for example, the center of the sigmoidal region on the DSC plot (Chapter 3). The difference in energy between the rigid and rubbery states corresponds to the increased amount of molecular motion experienced by the polymer after undergoing the glass transition. Below the glass transition temperature, the molecular motions are primarily vibrational, permitting little flexibility. Above the glass transition temperature, coordinated molecular motions occur, allowing the material to become rubbery. The higher the glass transition temperature, the more rigid the polymer, and the formation of a more rigid polymer is associated with greater force delivery, in other words with a higher value of elastic modulus.
Different orthodontic materials companies process their polyurethane products in different fashions. The two main methods of processing the modules are injection molding and die stamping. The die-stamped polymers have been found to be more consistent in physical properties. In addition, the polymer structure of the polyurethane modules may differ between companies (Figure 8.1). This is another source of variation in the glass transition temperature between different products, since the glass transition temperature is directly related to polymer structure. The primary structural influences on Tg are the composition, the steric interferences, and the types of bonding present in the molecule. All of these factors influence molecular motion.
An investigation by Renick and colleagues has shown that glass transition temperatures can differ among some polyurethane chain products by 15° −20 °C, as shown in Figures 8.2 and 8.3, indicating substantial differences in their structure or composition. (These DSC plots can be interpreted by referring to Fig. 3.12.) A higher Tg corresponds to a more rigid polymer, because of either the presence of more cross-linked chains or larger side-chains causing steric interferences. A general graph of the elastic modulus for a polymer as a function of temperature (Fig. 8.4) indicates the existence of a rubbery plateau region following the glass transition. A higher value of Tg may indicate a polymer that is in an earlier stage of the rubbery plateau at room temperature.
Pigmenting these products, which is done by manufacturers to increase patient acceptance, raises the question whether the pigments affect the structure of the elastomers. Certain types of fillers have been incorporated by the polymer industry to provide color. However, the compositions of the orthodontic chains are proprietary information, and information about the pigmentation process is not available. Whether the filler bonds to the polymer remains a question for future research. If the pigment can form cross-links with the polymer, it could potentially raise the glass transition temperature.
The study by Renick and colleagues of as-received products indicated that, while the use of gray, red, and purple pigments had no significant effect on the Tg for two brands, there was a significant difference in Tg for the gray-pigmented and purple-pigmented chains of the third brand. The DSC plots for specific brand-pigment combinations were similar for the as-received condition and in vitro testing in which four-segment modules were stretched for four weeks in air to a length of 2.5 cm, approximating the in vivo molar-to-canine distance. The DSC analyses of specimens after four weeks of in vivo use in randomly selected patients undergoing orthodontic treatment revealed a second glass transition (Fig. 8.5) that was not present for specimens in the as-received condition or after in vitro testing in air. The origin of this peak, which is due to the effects of the oral environment on the polyurethane polymer, is presently unknown. In general, studies focusing on the mechanical characteristics of colored elastomerics have failed to reveal differences between the conventional gray or clear elastomers and their pigmented counterparts.
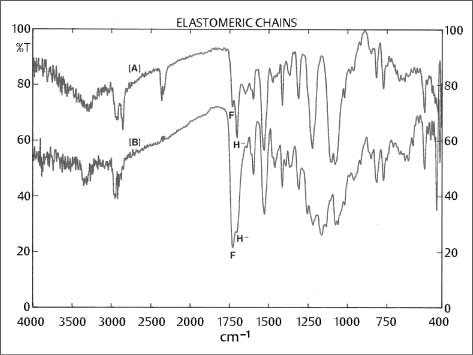
Fig. 8.1 Muliple internal reflectance FTIR spectra of two commercially available elastomeric chains [A] and [B], demonstrating different molecular composition and different extent of free (F) (1730 cm−1) vs. hydrogen-bonded (H-) (1730 cm−1) polyurethane carboxyls
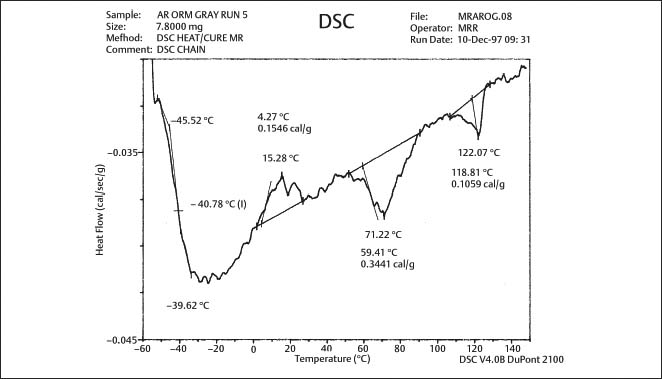
Fig. 8.2 DSC plot for as-received gray-pigmented elastomeric chain manufactured by Ormco. The glass transition temperature is approximately −41 °C (From Renick, 1999. An abstract describing this study is found in Renick et al, 1999a)
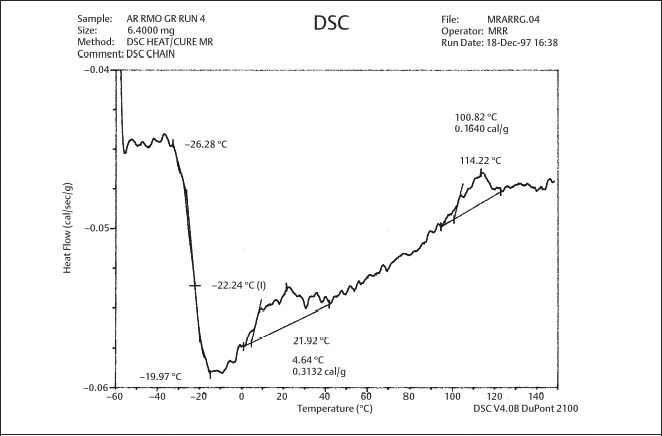
Fig. 8.3 DSC plot for as-received gray-pigmented elastomeric chain manufactured by RMO. The glass transition temperature is approximately −22 °C (From Renick, 1999. An abstract describing this study is found in Renick et al, 1999a)
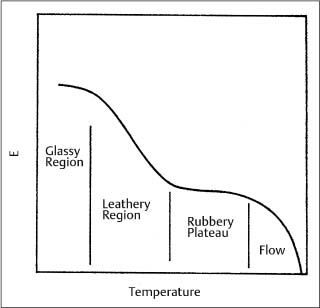
Fig. 8.4 Generalized dependence of the elastic modulus (E) for a polymer upon temperature, showing the trend in values for the different types of structures. (Adapted from Rosen, 1993)
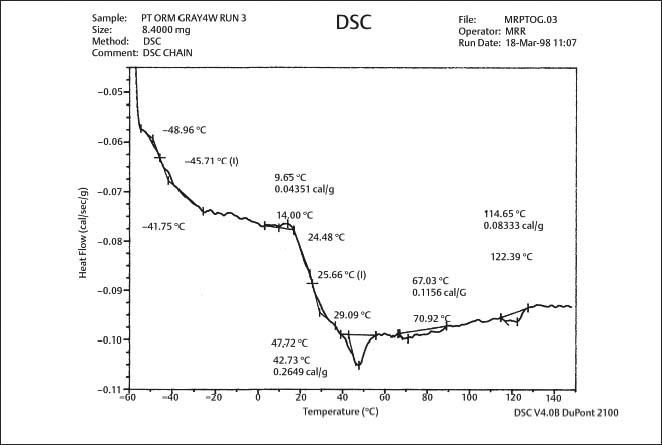
Fig. 8.5 DSC plot for gray-pigmented elastomeric chain manufactured by Ormco, following four weeks of use in vivo. Note the second glass transition at approximately 26 °C that is not present in Figure 8.2. (From Renick, 1999. An abstract describing this study is found in Renick et al, 1999b)
The general properties of elastomers, which follow from these preceding considerations, have been summarized by Billmeyer:
• When stretched rapidly, elongations greatly in excess of 100% can be achieved, with no major loss of energy.
• The highest values of tensile strength and stiffness are obtained after full stretching.
• Upon removal of the tensile force, a rapid contraction occurs, since the polymer structure has a strong tendency to return to its original condition.
• Full recovery takes place as long as the tensile force does not exceed the elastic limit, demonstrating the high resilience of these materials.
The form of the stress-strain curve for an elastomer and the dependence of force delivery and recovery on the rate of loading have been described in Chapter 1.
Elastomeric Ligatures
Conventional Ligatures
Elastomeric ligatures constitute the major medium engaging the archwire to a bracket slot. The clinician may prefer the use of these materials over the 0.20 mm to 0.36 mm (0.008–0.014 inch) stainless steel ligature wires for several reasons, including their ease of application, patient-friendly nature, aesthetic appearance, potential for fluoride release, and decreased fo/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


