chapter 6
The Various Presentations of Pain
The genesis of pain is poorly understood, if, indeed, it is understood at all. There was a time when it was generally thought that noxious stimulation, meaning tissue-damaging stimulation, was the chief basis for nociception. It was presumed that pain was some kind of gauge of tissue injury: the greater the pain, the greater the injury that had been sustained. Pain occurring without an initiating antecedent was presumed to be generated by psychic phenomena and therefore was not really pain at all.
As the concept of pain modulation evolved, the question of genesis lost much of its significance. It is now recognized that factors other than noxious stimulation are determinants of discomfort. For example, Wall1 has shown that the degree of pain experienced at the time of injury does not relate to the amount of tissue damage but instead to the attention given the injury. After the initial experience of pain associated with the tissue injury, the amount of continued pain and suffering more closely relates to the consequence of the injury and its treatment. Algogenic substances that accompany inflammation incidental to tissue injury require time to develop and therefore are not immediate determinants in the matter of pain from tissue damage per se. So, structural injury from mechanical, chemical, or thermal trauma, as it relates to the genesis of pain, abounds in uncertainty.
Not All Pains Are Alike
The word pain has different meanings to different individuals. Certainly the clinician who attempts to understand and manage pain needs to have a thorough appreciation for the different types of pain that can be encountered. Several types of pains will now be contrasted.
Clinical Versus Experimental Pain
Beecher2 pointed out that pain as presented by patients—so-called clinical or pathologic pain—is different from experimental pain induced and studied in the laboratory. The difference is illustrated in the capacity of morphine to give relief. A large dose of morphine does not significantly alter the brief jabs of experimental pain, while a much smaller dose consistently reduces pain that has meaning to the patient. The difference is also borne out in placebo effects. Beecher determined that the average effectiveness of placebos when dealing with clinical pain was about 35%, whereas it was only 3.2% with experimental pain. Nyquist and Eriksson, 3 in commenting on this problem, said, “We interpret our findings to suggest that clinical and experimental pain are explained by different mechanisms and that direct correlations cannot be drawn between them.” Bromm4 believed that experimental pain was actually a measurement of the sensory-discriminative component only, not the aversive-emotional component that is displayed in clinical pain.
It is likely that the emotional significance embodied in clinical pain is the real determinant. Indeed, the purpose of the signal detection (sensory decision) theory for determining the effectiveness of analgesic medications is to minimize this bias factor.5,6
Acute Versus Chronic Pain
As the duration of pain input continues, the level of suffering increases even though the intensity of the input remains the same. In fact, protracted input may result in a high level of discomfort, even if the intensity decreases or the input disappears altogether. Perhaps this increase in pain intensity in the presence of less peripheral nociceptive input may result from a decrease in the effectiveness of the descending inhibitory system (see chapter 4). It follows that a sustained level of discomfort may remain even after the initiating cause has resolved. With chronicity, all pains, regardless of initial type or origin, seem to take on the clinical characteristics of psychologic intensification. As such, some chronic orofacial pain syndromes display features suggestive of depressive illness.7,8,9 Treatment measures effective for alleviating acute pain may no longer be applicable. As pain becomes chronic, management options shift from local to systemic modalities. Pain that initially could be managed on a purely dental level may require extensive and coordinated interdisciplinary therapy when it becomes chronic.
It should be noted that there is a definite relationship between intensity and duration of pain. The higher the intensity, the shorter the period that can be tolerated by the sufferer. Low-intensity pain can be sustained for up to 7 hours, whereas maximum-intensity pain can be tolerated for no more than a few seconds. The higher the intensity, the more likely it is that the pain will be intermittent. Only low-intensity pain can be protracted. Intractable pain, regardless of the patient’s contention to the contrary, must be either periodic or of extremely low intensity. In fact, intractable pain may be little more than an unpleasant or unwanted sensation.10,11(pp3–28),12
Normal Neural Activities: Central and Peripheral Mechanisms
With the discovery of the normal ongoing barrage of afferent signals generated in the dorsal root ganglia and the countered action of the brainstem descending inhibitory mechanism, a better concept of pain inception and accentuation has developed. It is known that peripheral nerve injury accentuates the normal ongoing barrage of afferent signals.13 Likewise, it is known that nonpainful sensation conducted by the superficial cutaneous nerves effectively accentuates the inhibitory activity of the descending mechanism, thus shifting the balance away from painful sensation experience. This activity has been more broadly called the diffuse noxious inhibitory control14 (see chapter 4). There is good reason to presume that most, if not all, exteroceptive sensory input, including that of the special senses, has a similar counterbalancing effect on the dorsal ganglion source of sensory impulses.
The important issue here is that central mechanisms may actually be the chief factor in the inception of pain, rather than any peripheral input. Indeed, pain may be felt in the absence of noxious stimulation when the descending inhibitory mechanism is deficient, when the ganglionic barrage is accentuated, or both. Likewise, little or no pain may be sensed from otherwise painful causes when the descending mechanism more effectively acts to arrest the passage of impulses to the higher centers. It is known that both peripheral and central influences affect the action of this inhibitory mechanism. 14,15,16
There are many possibilities, both peripheral and central, that may exert an influence on what is felt as pain. We are just beginning to learn the significance of these factors. The antinociceptive action of superficial stimulation (for example, pressure, massage, analgesic balms, vibration, thermal applications, hydrotherapy, counterirritation, transcutaneous electrical nerve stimulation) is quite well known. The inhibitory effect of distraction, physical activity, and calm serenity as contrasted with the excitatory effect of attention, fear, anxiety, and consequences very likely also involves activity of the descending inhibitory mechanism and the diffuse noxious inhibitory control.
With the discovery of the axon transport system and the antidromic passage of neurotransmitters and other neurochemicals peripherally, a better understanding of the importance of the neurogenic component of inflammation is developing (see chapter 4). Since cutaneous effects of wheals (edema) and flare (hyperemia) are now known to result from such antidromic activity, 17 the intimate relationship between neural activities and inflammation makes it difficult to decide what part of an inflammatory process generates pain and what part is actually the result of pain. In other words: Is the muscle painful because of local tissue reasons such as overuse, or is the central nervous system (CNS) responsible for the muscle soreness by way of antidromic neurogenic inflammation? This is an important question that must be appreciated and understood by the clinician, since the treatment of these two conditions is very different.
Inflammatory Pain
The traditional concept of noxious stimulation as the initiating origin of pain stems from an inflammatory process. Tissue injury initiates an inflammatory reaction that characteristically induces pain. The symptoms relate to the conditions that prevail, such as: (1) the kind, extent, and location of injury; (2) the reactivity of the injured structures; (3) the degree of confinement of the inflammatory exudate; and (4) the phase of inflammation.
Inflammatory pain is due chiefly to the action of prostaglandins and bradykinin, substances released by the inflammatory process. They act in conjunction with each other to increase local vasodilation and capillary permeability as well as alter the sensitivity and receptivity of receptors in the area18,19,20,21,22,23 (see chapter 4). Thus, the pain threshold is lowered so that nociceptors become more sensitive to stimulation, and higher-threshold mechanoreceptors are sensitized to a wider variety of stimuli. As a result, spontaneous primary pain and stimulation-evoked primary hyperalgesia take place. Along with these local effects, there is a prostaglandin-like substance released in the CNS that sensitizes nociceptive interneurons to mechanical and chemical stimuli and renders neural pathways related to inflammatory pain more sensitive to the action of opiates. This has been called prostaglandin hyperalgesia.24
Pain of inflammatory origin may involve different kinds of tissue innervated by receptors with different reactive responses. For example, superficial pain may be inflammatory, such as that of dermatitis or gingivitis. Examples of musculoskeletal inflammatory pain are those of myositis and cellulitis. Visceral inflammatory pain may be expressed as that of lymphadenitis or arteritis. Inflammatory neuropathic pain may be manifested as neuritis. Many physical ailments are inflammatory, and pain of the inflammatory type is part of the symptom complex.
It should be noted that inflammation due to local tissue injury is a normal immune response that initiates the healing process.25 The pain generated by inflammation functions to alert the subject to its presence. This type of pain exerts a protective inhibitory influence on biomechanical activity, and it serves to monitor the progress toward recovery of the injured part. Inflammation therefore should be looked upon as a protective and beneficial body function. Failure of the immune system to respond appropriately invites disaster. Artificial suppression of the inflammatory process to minimize pain may be counterproductive therapeutically. The recognition of inflammatory pain should alert the examiner to the need for cause-related therapy.
Noninflammatory Pain
It is likely that noninflammatory pain is a much more common source of suffering than inflammatory pain. This concept, however, is certainly not the traditional approach to explaining pain. Most clinicians have been trained to establish a diagnosis that ends in “itis” (eg, tendonitis, gastritis, otitis). This suggests that the tissue or organ described is inflamed. This is certainly not true of many pain disorders. By this time it is hoped that the reader has an appreciation for the many conditions that exist in the somatic tissues and CNS that may create pain in the absence of inflammation.
It is useful to diagnostically separate inflammatory pains from noninflammatory pains because of their different clinical presentations and courses. As previously stated, true tissue inflammation is a result of an immune response that causes certain changes in local tissues, such as reddening, increased temperature, and edema. Inflammation follows a relatively predictable time course, getting worse and then, with healing (with or without therapy), getting better. Most pains do not show these characteristics, at least not at the clinical level, and are therefore considered to be noninflammatory pains. For example, in this text neurogenic inflammation will not be considered in the category of a true inflammatory disorder, since it does not seem to present with the obvious local clinical symptoms associated with inflammation. It is acknowledged that this separation may not be distinguishable or appropriate; however, it does offer some diagnostic advantages.
Noninflammatory pain may originate from somatic (soma is Latin for body) or neurogenous structures. Some of the more common disorders and features of these two categories will be presented here, with a more complete description to follow in future chapters.
Somatic Pain
Somatic pains are pains that originate from body structures and may be divided into two main types: superficial and deep.
Deep somatic pains
Deep somatic pains originate from all of the structures that are located deep within the envelope, with the exception of the neural structures. Deep somatic structures are either musculoskeletal structures or visceral structures (the supply system). Within these two categories fall some of the most common pains of human suffering, specifically, muscle pains and vascular pains.
Musculoskeletal pains
Noninflammatory muscle pain is the most common type of somatic pain, and yet it remains very poorly understood. A great deal of mystery continues to shroud myogenous pain in general; this affects clinicians and patients alike. Without a good understanding of this pain one cannot effectively manage the patient’s complaints. Muscle pain may frequently occur in relationship to systemic responses. Some pains are a result of a protective response called co-contraction (formally called muscle splinting). Co-contraction may result from any alteration in sensory or proprioceptive input. It also may be a response to deep pain or even the threat of injury. If this condition is protracted, delayed-onset muscle soreness (or local muscle soreness) may result.
Another painful muscle condition is myofascial trigger point pain. This type of pain is characterized by localized areas of hypersensitive bands of muscle fibers called trigger points. These trigger points often produce heterotopic pains that dominate the patient’s complaints.
Occasionally muscle pain may originate from a combination of peripheral factors and the CNS. This may induce contraction of the entire muscle, called a myospasm. When muscle pain becomes protracted, myositis may dominate in the clinical presentation. True muscle inflammation, however, is rare unless associated with trauma or other injury.
An additional type of muscle pain may originate solely from the CNS. This condition is termed centrally mediated myalgia and is dependent on the neurogenic inflammation produced antidromically by the central mechanisms.
Musculoskeletal pains also include the articular surfaces of joints and their associated structures. Various types of arthritides can be a source of deep pain. When discussing orofacial pains, the temporomandibular joint becomes the joint of interest.
Visceral pains
Visceral pains originate from tissues associated with the supply system. Structures that are of concern in the orofacial region are visceral mucosa (sinus), tooth pulps, and glandular, ocular, and auricular structures. Vascular tissues of the orofacial structures may become painful secondary to inflammation. Such conditions are called vasculitis and arteritis. The greatest source of vascular pain, however, comes from neurogenic inflammation of the vessel walls. Since the origin of the neurogenic inflammation is the afferent nerve that innervates the vessel wall, this type of pain is termed neurovascular pain, also called migraine. Migraine may likely represent one of the most common pain complaints known to humans.26 Unfortunately, vascular pain continues to be poorly understood by health professionals. Gross27 has shown that blood vessels are innervated by afferent neurons of both the somatic and visceral nervous systems. Nociceptive impulses arising from vascular structures therefore may be mediated by either or both of these pathways.
It was originally thought that neurovascular pain was initiated chiefly by dilation of small arteries, which distorted and noxiously stimulated afferent fibers in vascular and perivascular tissues. Vasoconstricting medications such as ergotamine tartrate, which reduce the dilation and amplitude of pulsation of arteries, decrease the pain. Pressure applied to the carotid artery also diminishes the pain.11(pp3–28) The presumption, however, that vasodilation is the primary cause of neurovascular pain is no longer tenable, because spontaneous dilation of cranial arteries does not cause pain in normal subjects. Rather, dilating drugs provide relaxation of arteries but do not cause pain. Only algogenic substances such as bradykinin and histamine can evoke vasodilation and pain.28 It is more likely that a neurogenic inflammation within the trigeminal neurovascular complex is responsible for this type of neurovascular pain (see chapter 16).
Systemic factors, more than local factors, seem to play the major role in the incidence of neurovascular pain. These pains seem to occur more commonly in certain “pain patients,” called hypernoceptors, who are thought to have some deficiency in opioid peptides and neurotransmitters of the antinociceptive system.28 Serotonin deficiency in the CNS has been identified with the incidence of neurovascular pain.29,30,31,32 Administration of histamine to known “migraine patients” consistently precipitates an attack of pain, while nonmigrain-ous persons do not react in this way.33 Patients with neurovascular pain frequently display a characteristic emotional overlay with personality features of insecurity with tension.34 This may be manifested as inflexibility, conscientiousness, meticulousness, perfectionism, and resentment. Frustration, fatigue, and prostration seem to set the stage for an attack.11(pp3–28) In the light of serotonin depletion, which also characterizes depressive states,35,36 the emotional symptoms and the pain may simply represent different facets of a common problem. 37,38,39
Early on, Wolff11(pp3–28) described four conditions that seemed to contribute to the discomfort of neurovascular pain: (1) dilation of blood vessels, (2) local edema of the painful site, (3) edema of the vessel wall and perivascular tissue, and (4) associated muscle pain (especially in the occipital area). It has been shown that attacks of migraine without aura (see chapter 16) display accompanying increased electromyographic activity in pericranial muscles.40,41
It appears that tension-type headache (commonly associated with myofascial pain) and neurovascular headache (especially migraine without aura) have much in common.28,42 The clinical complaint frequently comprises elements of both varieties of pain. It is likely that the systemic factors in these conditions stem from a common cause, and the peripheral symptoms may represent little more than different facets of a common problem. The psychologic implications of both conditions therefore are of great importance.
Since neurovascular pain appears to be originating in the vessel, it is perfectly logical to consider this a type of visceral pain. However, recent information alters our original view of this most common and complex type of pain. It appears that the phenomenon of neurovascular pain begins with a triggering of a trigeminal neurovascular mechanism. Since the initiating factor appears to be associated with neurogenic mechanism, the classification of migraine will be discussed in this text under the category of neurovascular pain disorders (see chapter 16).
Neuropathic Pain
Some clinical pains arise from abnormalities in the neural structures themselves and are therefore called neuropathic pains. Neuropathic pains can be confusing to the clinician, since the clinical examination in the area of the reported pain reveals no somatic tissues changes. The clinical examination reveals no reason for the pain condition. The novice clinician is likely to jump to the conclusion that this is a psychogenic pain. An understanding of neuropathic pain is fundamental in the management of orofacial pains since it is common in the head and neck. These pains may be divided into two groups, according to their presentation: paroxysmal neuropathic pains and continuous neuropathic pains.
Paroxysmal neuropathic pains
Some neuropathic pains have a clinical course that reveals very high and sudden levels of pain, followed by periods of total remission of symptoms. These sudden electrical-like volleys of pain are typically felt in the exact distribution of the involved nerve. These shock-like pains are called paroxysmal pains and are usually momentary and severe. They are often triggered by relatively mild mechanical stimulation at a site in the peripheral distribution of the same nerve involved in the neuralgia. The pains are often recurrent, with no pain experienced between the episodes. Paroxysmal neuralgias are named for the nerve that is affected, for example, trigeminal neuralgia (originally known as tic douloureux).
Paroxysmal trigeminal neuralgia is one of the most painful conditions known to humans, yet it remains an enigma to many health professionals. It is thought that paroxysmal neuralgic pains are closely associated with demyelination of the nerve fiber. Myelination of peripheral neurons helps to isolate the transmission of impulses within the single neuron. Transfer of signals from one neuron to another normally takes place only at synapses. If the myelin sheath is lost, however, neural impulses can be transferred to a neighboring neuron. Kerr43 observed that demyelination of peripheral neurons can lead to the ephaptic transmission of impulses. (An ephapse is a point of lateral contact, other than a synapse, between nerve fibers across which impulses are conducted directly through the nerve membranes from one fiber to the other.) Experimental demyelination leads to antidromically propagated extraction potentials, as well as some post-discharge activity followed by refractory periods. 44 Short-term peripheral nerve compression is usually painless and rarely lasts more than a few seconds.45,46 Continued compression causes local demyelination without a loss of axonal continuity. Chronic nerve entrapment causes demyelination initially, followed by progressive axonal degeneration. In traumatic neuroma, ephaptic cross-excitation may be a factor in the generation of pain.47
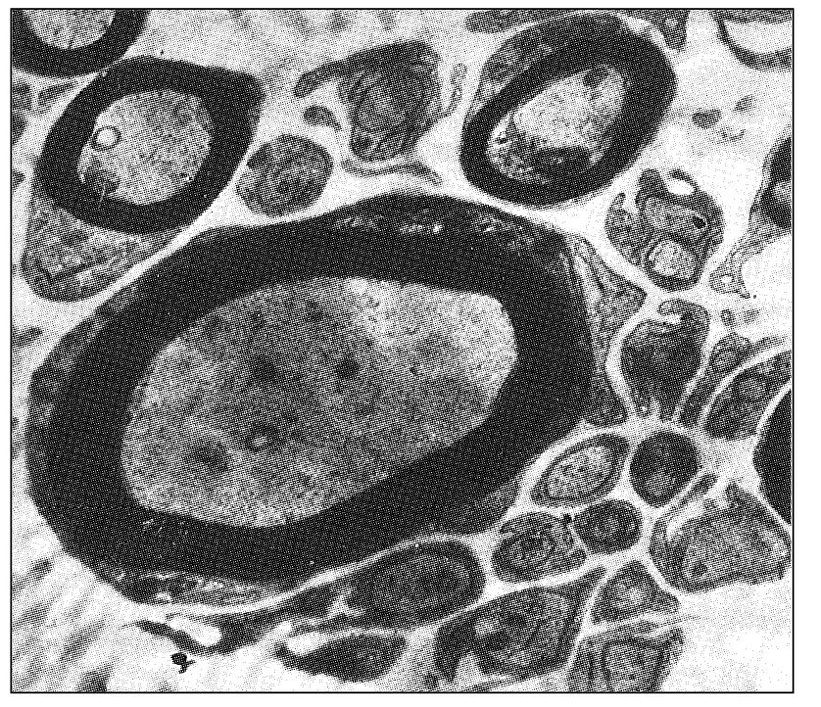
Fig 6-1 Transverse section through the posterior rootlet of the human trigeminal nerve that illustrates an area of unusually abundant unmyelinated fibers and several myelinated fibers (original magnification × 10,000). (From Kerr FWL. Fine structure and functional characteristics of the primary trigeminal neuron. In: Hassler R, Walker AE (eds). Trigeminal Neuralgia: Pathogenesis and Pathophysiology. Stuttgart: Thieme, 1970. Used with permission.)
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


