Biocompatibility and Tissue Reaction to Biomaterials
Biocompatibility is formally defined as the ability of a material to elicit an appropriate biological response in a given application in the body. Inherent in this definition is the idea that a single material may not be biologically acceptable in all applications. For example, a material that is acceptable as a full cast crown may not be acceptable as a dental implant. Also implicit in this definition is an expectation for the biological performance of the material. In a bone implant, the expectation is that the material will allow the bone to integrate with the implant. Thus an appropriate biological response for the implant is osseointegration. In a full cast crown, the expectation is that the material will not cause inflammation of pulpal or periodontal tissues, but osseointegration is not an expectation. Whether or not a material is biocompatible therefore depends on the physical function for which the material will be used and the biological response that will be required from it. Using this definition, it makes little sense to say that any given material is or is not biocompatible—how the material will be used must be defined before that can be assessed.
In that regard, biocompatibility is much like color. Color is a property of a material interacting with its environment (light), and the color of a material depends on the light source and the observer of the light. Similarly, biocompatibility is a property of a material interacting with its environment. The biological response will change if changes occur in the host, the application of the material, or the material itself (Figure 6-1).
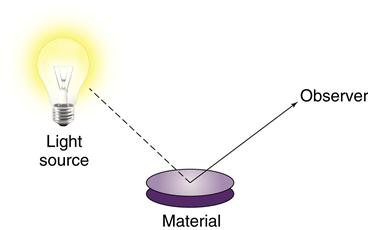
Like color, biocompatibility is not a property of just a material, but rather a property of how a material interacts with its environment. A material’s color depends on the character of the light source, how the light interacts with the material, and how the observer interprets the reflected light. In this sense, the material’s color depends on its environment. The biocompatibility of a material is similar in the sense that it depends on its environment as well as the nature of the material.
Dentistry shares concerns about biocompatibility with other fields of medicine, such as orthopedics, cardiology, and vascular biology, among others. In the development of any biomaterial, one must consider the strength, esthetics, and functional aspects of the material, as well as its biocompatibility. Furthermore, demands for appropriate biological responses are increasing as materials are expected to perform more sophisticated functions in the body for longer time periods. Thus considerations of biocompatibility are important to manufacturers, practitioners, scientists, and patients. The field of biocompatibility is interdisciplinary and draws on knowledge from materials science, bioengineering, biochemistry, molecular biology, tissue engineering and other fields.
This chapter briefly surveys the tests used for evaluating biocompatibility of dental materials and how well they correlate with one another, overviews the specifications that govern such testing, and describes the strengths and weaknesses of the testing methods. The majority of the chapter is devoted to a discussion of the biocompatibility of the various materials used in dentistry within the framework of these principles.
Measuring Biocompatibility
Measuring the biocompatibility of a material is not simple, and the methods of measurement are evolving rapidly as more is known about the interactions between dental materials and oral tissues and as technologies for testing improve. Historically, new materials were simply tested in humans to assess their biocompatibility. However, this practice has not been acceptable for many years, and current materials must be extensively screened for biocompatibility before they are used in humans. Several varieties of tests are currently used to ensure that new materials are biologically acceptable. These tests are classified as in vitro, animal, and usage tests. These three testing types include clinical trials, which is really a special case of a usage test in humans. This section discusses examples of each type of test, their advantages and disadvantages, how the tests are used together, and standards that rely on these tests to regulate the use of materials in dentistry.
In Vitro Tests
In vitro tests for biocompatibility, done outside a living organism, require placement of a material or a component of a material in contact with a cell, enzyme, or some other isolated biological system. The contact can be either direct, when the material contacts the cell system without barriers, or indirect, when there is a barrier of some sort between the material and the cell system. Direct tests can be further subdivided into those in which the material is physically present with the cells and those in which some extract from the material contacts the cell system. In vitro tests can be roughly subdivided into those that measure cytotoxicity or cell growth, those that measure some metabolic or other cell function, and those that measure the effect on the genetic material in a cell (mutagenesis assays). Often there is overlap in what a test measures. In vitro tests have a number of significant advantages over other types of biocompatibility tests (Table 6-1). They are relatively quick, generally cost less than animal or usage tests, can be standardized, are well suited to large-scale screening, and can be tightly controlled to address specific scientific questions. The overriding disadvantage of in vitro tests is their questionable relevance to the final in vivo use of the material (see later section on correlation between tests on page 114). Another significant disadvantage includes the lack of inflammatory and other tissue-protective mechanisms in the in vitro environment. It should be emphasized that in vitro tests alone cannot entirely predict the overall biocompatibility of a material.
TABLE 6.1
Advantages and Disadvantages of Biocompatibility Tests
| Test | Advantages | Disadvantages |
| In vitro tests | Quick to perform | Relevance to in vivo is questionable |
| Least expensive | ||
| Can be standardized | ||
| Large-scale screening | ||
| Good experimental control | ||
| Excellence for mechanisms of interactions | ||
| In vivo tests | Allows complex systemic interactions | Relevance to use of material is questionable |
| Response more comprehensive than in vitro tests | Expensive | |
| More relevant than in vitro tests | Time consuming | |
| Legal/ethical concerns | ||
| Difficult to control | ||
| Difficult to interpret and quantify | ||
| Usage tests | Relevance to use of material is assured | Very expensive |
| Very time consuming | ||
| Major legal/ethical issues | ||
| Can be difficult to control | ||
| Difficult to interpret and quantify |
Standardization of in vitro tests is a primary concern. Two types of cells can be used for in vitro assays. Primary cells are those cells taken directly from an animal and cultured. These cells will grow for only a limited time in culture but usually retain many of the characteristics of cells in vivo. Continuously grown cells or cell lines are cells that have been transformed previously to allow them to grow more or less indefinitely in culture. Because of this transformation, these cells do not retain all in vivo characteristics, but they do consistently exhibit those features that they do retain. Primary cell cultures seem to be more relevant than continuous cell lines for measuring cytotoxicity of materials. However, primary cells, being from a single individual, have limited genetic variability, may harbor viral or bacterial agents that alter their behavior, and often rapidly lose their in vivo functionality once placed in culture. Furthermore, the genetic and metabolic stability of continuous cell lines contributes significantly toward standardizing assay methods. In the end, both primary and continuous cells both play important roles in in vitro testing; and both should be used to assess materials.
Cytotoxicity Tests
Cytotoxicity tests assess cell death caused by a material by measuring cell number or growth before and after exposure to that material. Control materials should be well defined and commercially available to facilitate comparisons among other testing laboratories. Membrane permeability tests are used to measure cytotoxicity by the ease with which a dye can pass through a cell membrane, because membrane permeability is equivalent to or very nearly equivalent to cell death (Figures 6-2 and 6-3).
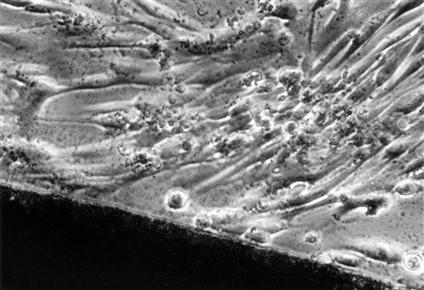
Light microscopic view of a noncytotoxic interaction between a material (dark image at bottom of the picture) and periodontal ligament fibroblasts in a cell culture (in vitro) test. The morphology of the fibroblasts indicates that they are alive and are not suffering from a toxic response (see Figure 6-3 for contrast). The material in this case was a calcium hydroxide pulp-capping agent.
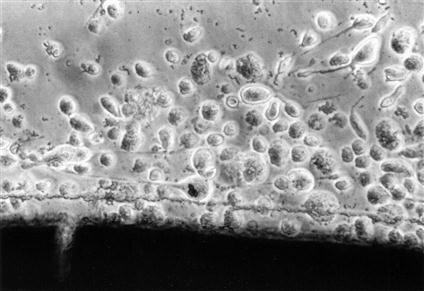
Light microscopic view of a cytotoxic interaction between a material (dark image at the bottom of the picture) and periodontal ligament fibroblasts in a cell culture test. The fibroblasts are rounded and detached (see Figure 6-2 for contrast), indicating that they are either dead or dying. The material is a type of calcium hydroxide pulp-capping agent, different from the one shown in Figure 6-2.
Tests for Cell Metabolism or Cell Function
Some in vitro tests for biocompatibility use the biosynthetic or enzymatic activity of cells to assess cytotoxic response. Tests that measure deoxyribonucleic acid (DNA) synthesis or protein synthesis are common examples of this type of test. A commonly used enzymatic test for cytotoxicity is the MTT (MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] test, as well as the NBT (nitroblue tetrazolium), XTT [2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt], and WST (a water-soluble tetrazolium), all being colorimetric assays based on different tetrazolium salts. Alamar Blue tests quantitatively measure cell proliferation using a fluorescent indicator that allows continuous monitoring of cells over time.
Tests That Use Barriers (Indirect Tests)
Because direct contact often does not exist between cells and materials during in vivo use, several in vitro barrier tests have been developed to mimic in vivo conditions. These tests include an agar overlay method, which uses agar to form a barrier between the cells and the material, and the Millipore filter assay, in which a monolayer of cells is grown on a filter that is turned over so that test materials are placed on the filter and leachable diffusion products are allowed to interact with the cells. The agar diffusion and Millipore filter tests can provide, at best, a qualitative cytotoxic ranking among materials (Figure 6-4).
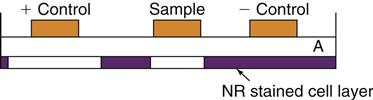
The agar overlay method has been used to evaluate the cytotoxicity of dental materials. The cell layer, which has been previously stained with neutral red (NR), is covered with a thin layer of agar (A). Samples are placed on top of the agar for a time. If the material is cytotoxic, it will injure the cells and the neutral red will be released, leaving a zone of inhibition.
A number of studies have shown that dentin forms a barrier through which toxic materials must diffuse to reach pulp tissue, with the thickness of the dentin directly correlating with the protection offered to the pulp. These assays, which incorporate dentin disks between the test sample and the cell assay system, have the added advantage of directional diffusion between the restorative material and the culture medium (Figure 6-5).
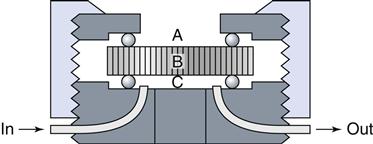
A dentin disk is used as a barrier in cytotoxicity tests that attempt to predict the toxicity of materials placed on dentin in vivo. The material is placed on one side (A) of the dentin disk (B) in the device used to hold the dentin disk. Collection fluid (cell culture medium or saline) is on the other side of the disk (C). Cells can also be grown on the collection side. Components of the material may diffuse through the dentin and the effect of the medium on cell metabolism can then be measured. To assess the rate of diffusion, the collection fluid can be circulated into and out of the collection chamber (C).
Other Assays for Cell Function
In vitro assays to measure immune function or other tissue reactions have also been used. These assays measure cytokine production by lymphocytes and macrophages, lymphocyte proliferation, chemotaxis, or T-cell rosetting to sheep red blood cells. Other tests measure the ability of a material to alter the cell cycle or activate complement. The in vivo significance of these assays is yet to be ascertained, but many show promise for being able to reduce the number of animal tests required to assess the biocompatibility of a material.
Mutagenesis Assays
Mutagenesis assays assess the effect of a biomaterial on a cell’s genetic material. There are a wide range of mechanisms by which a material can affect a cell’s genes. Genotoxic mutagens directly alter cell DNA through various types of mutations. Each chemical may be associated with a specific type of DNA mutation. Genotoxic chemicals may be mutagens in their native states, or may require activation or biotransformation to be mutagens, in which case they are called promutagens. Epigenetic mutagens do not alter the DNA themselves, but support tumor growth by altering the cell’s biochemistry, altering the immune system, acting as hormones, or by other mechanisms. Carcinogenesis is the ability to cause cancer in vivo. Mutagens may or may not be carcinogens, and carcinogens may or may not be mutagens. Thus quantitation and relevance of tests that measure mutagenesis and carcinogenesis are extremely complex. A number of government-sponsored programs evaluate the ability of in vitro mutagenesis assays to predict carcinogenicity.
The Ames test is the most widely used short-term mutagenesis test and the only short-term one that is considered thoroughly validated. It looks at the conversion of a mutant stock of Salmonella typhimurium back to a native strain, because chemicals that increase the frequency of reversion back to the native state have a high probability of being carcinogenic in mammals. A second test for mutagenesis is the Styles’ cell transformation test. This test on mammalian cells offers an alternative to bacterial tests (Ames test), which may not be relevant to mammalian systems. However, because the Ames test is widely used, extensively described in the literature, and technically easier to conduct in a testing laboratory than the other test, it is most often conducted in a screening program.
Animal Tests
Animal tests for biocompatibility, usually involving mammals such as mice, rats, hamsters, or guinea pigs, are distinct from usage tests (which are also often done in animals) in that the material is not placed in the animal with regard to its final use. The use of an animal allows for the complex interactions between the material and a functioning, complete biological system to occur. This is extremely difficult to mimic in a cell-culture system. Thus the biological responses in animal tests are more comprehensive and may be more relevant than in vitro tests, and these features are the major advantages of these tests (see Table 6-1). The main disadvantages of animal tests are that they can be difficult to interpret and control, are expensive, time consuming, and often involve significant ethical concerns and oversight. Furthermore, the relevance of the test to the in vivo use of a material is often unclear, especially in estimating the appropriateness of an animal species to represent a human. A variety of animal tests have been used to assess biocompatibility.
The mucous membrane irritation test determines whether a material causes inflammation to mucous membranes or abraded skin. In a skin sensitization test, materials are injected intradermally to test for development of skin hypersensitivity reactions, followed by secondary treatment with adhesive patches containing the test substance. Implantation tests are used to evaluate materials that will contact subcutaneous tissue or bone. The location of the implantation site is determined by the use of the material and may include connective tissue, bone, or muscle. Although amalgam and alloys are tested because the margins of the restorative materials contact the gingiva, most subcutaneous tests are used for materials that will directly contact soft tissue during implantation, as well as endodontic and periodontal treatment materials.
Usage Tests
Usage tests may be done in animals or in human study participants. They are distinct from other animal tests because they require that the material be placed in a situation identical to its intended clinical use. The usefulness for predicting biocompatibility is directly proportional to the fidelity with which the test mimics the clinical use of the material in every regard, including time, location, environment, and placement technique. For this reason, usage tests in animals usually employ larger animals that have similar oral environments to humans, such as dogs, mini-swine or monkeys. When humans are used, the usage test is termed a clinical trial. The overwhelming advantage for usage tests is their relevance (see Table 6-1). These tests are the gold standard, in that they give the ultimate answer to whether or not a material will be biocompatible and clinically useful. One might ask, then, why bother with in vitro or animal tests at all. The answer is in the significant disadvantages of the usage test. These tests are extremely expensive, last for long periods, involve many ethical and often legal concerns, are exceptionally difficult to control and interpret accurately, and may harm the test participants. Additionally, statistical analysis of these tests is often a daunting process. In dentistry, dental pulp, periodontium, and gingival or mucosal tissues are the main targets of usage tests.
Dental Pulp Irritation Tests
Generally, materials to be tested on the dental pulp are placed in class 5 cavity preparations in intact, noncarious teeth. At the conclusion of the study, the teeth are removed and sectioned for microscopic examination, with tissue necrotic and inflammatory reactions classified according to the intensity of the response. Although most dental-pulp irritation tests have involved intact, noncarious teeth, without inflamed pulps, there has been increased concern that inflamed dental pulp tissue may respond differently than normal pulps to liners, cements, and restorative agents. So, usage tests on teeth with induced pulpitis, which allow evaluation of the type and amount of reparative dentin formed, will likely continue to be developed and refined.
Dental Implants in Bone
At present, the best predictors for success of implants are careful patient selection and ideal clinical conditions. The following terms are used to define various degrees of success: early implant success for implants surviving 1 to 3 years, intermediate implant success for implants surviving 3 to 7 years, and long-term success for implants surviving more than 7 years. As such, there are three commonly used tests to predict implant success: (1) penetration of a periodontal probe along the side of the implant, (2) mobility of the implant, and (3) radiographs indicating either osseous integration or radiolucency around the implant. A bone implant is considered successful if it exhibits no mobility, no radiographic evidence of peri-implant radiolucency, has minimal vertical bone loss and is completely encased in bone, and has an absence of persistent peri-implant soft-tissue complications. Any fibrous capsule formation is a sign of irritation and chronic inflammation, which is likely to lead to micromotion of the implant and ultimately to loosening and failure.
Mucosa and Gingival Usage Tests
Tissue response to materials with direct contact of gingival and mucosal tissues is assessed by placement in cavity preparations with subgingival extensions. The material’s effect on gingival tissues are observed and responses are categorized as slight, moderate, or severe, depending on the number of mononuclear inflammatory cells (mainly lymphocytes and neutrophils) in the epithelium and adjacent connective tissues. A difficulty with this type of study is the frequent presence of some degree of preexisting inflammation in gingival tissue due to the presence of bacterial plaque, surface roughness of the restorative material, open or overhanging margins, and over- or under-contouring of the restoration.
Correlation Among In Vitro, Animal, and Usage Tests
In the field of biocompatibility, some scientists question the usefulness of in vitro and animal tests in light of the apparent lack of correlation with usage tests and the clinical history of materials. However, lack of correlation is not surprising in light of differences among these tests. in vitro and animal tests often measure aspects of biological response that are more subtle or less prominent than those observed during a material’s clinical use. Furthermore, barriers between the material and tissues may exist in usage tests or clinical use, but may not exist in the in vitro or animal tests. Thus it is important to remember that each type of test has been designed to measure different aspects of biological response to a material, and correlation is not always to be expected.
The best example of a barrier that occurs in use but not during in vitro testing is the dentin barrier. When restorative materials are placed in teeth, dentin will generally be interposed between the material and the pulp. The dentin barrier, although possibly only a fraction of a millimeter thick, is effective in modulating the toxic effect of a dental material. This dentin barrier effect is illustrated by the following classic study (Table 6-2). Three methods were used to evaluate the following materials: ZOE (zinc oxide eugenol) cement; resin composite; and silicate cement. The evaluation methods included (1) four different cell culture tests, (2) an implantation test, and (3) a usage test in class 5 cavity preparations in monkey teeth. The results of the four cell culture tests were relatively consistent, with silicate having only a slight effect on cultured cells, composite a moderate effect, and ZOE a severe effect. These three materials were also embedded subcutaneously in connective tissue in polyethylene tubes (secondary test), and observations were made at 7, 30, and 90 days. Reactions at 7 days could not be determined because of inflammation caused by the operative procedure. At 30 days, ZOE caused a more severe reaction than silicate cement. The inflammatory reactions at 90 days caused by ZOE and silicate were slight, whereas the reaction to resin composites was moderate. When the three materials were evaluated in class 5 cavity preparations under prescribed conditions of cavity size and depth (usage test), the results were quite different from those obtained by the other methods. The silicate was found to have the most severe inflammatory reaction, the composite had a moderate to slight reaction, and the ZOE had little or no effect.
TABLE 6.2
Comparison of Reactions of Three Materials by Screening and Usage Tests
< ?comst?>
| Material | Cell Culture | Implantation in Connective Tissue | Pulp Response |
| Silicate | + | + | + + |
| Resin composite | + + | + + | + |
| ZOE | + + + | + | 0 |
< ?comen?>< ?comst1?>

< ?comst1?>
< ?comen1?>
+ + +, Severe; + +, moderate; +, slight; 0, no reaction.
ZOE, Zinc oxide–eugenol.
From Mjör IA, Hensten-Pettersen A, Skogedal O: Int. Dent. J. 27,127, 1977.
Apparent contradictions in this study are explained by considering the components that were released from the materials and the environments into which they were released. The silicate cement released hydrogen ions that were probably buffered in the cell culture and implantation tests but were not adequately buffered by the dentin in the usage tests. Microleakage of bacteria or bacterial products may have added to the inflammatory reaction in those usage tests. Thus this material appeared to be the most toxic in the usage test. The composites released low-molecular-weight resins, and the ZOE released eugenol and zinc ions. In the cell culture tests, these compounds had direct access to cells and probably caused the moderate to severe cytotoxicity. In the implantation tests, the released components may have caused some cytotoxicity, but the severity may have been reduced because of the capacity of the surrounding tissue to disperse the toxins. In usage tests, these materials probably were less toxic because the diffusion gradient of the dentin barrier reduced concentrations of the released molecules to low levels. The slight reaction observed with the composites may also have been caused in part by microleakage around these restorations. The ZOE did not show this reaction, however, because the eugenol and zinc probably killed bacteria in the cavity, and the ZOE may have reduced microleakage.
Another example of the lack of correlation of usage tests with implantation tests is the inflammatory response of the gingiva at the gingival and interproximal margins of restorations that accumulate bacterial plaque and calculus. Plaque and calculus cannot accumulate on implanted materials and therefore the implantation test cannot hope to duplicate the usage test. However, connective tissue implantation tests are of great value in demonstrating the cytotoxic effects of materials and evaluating materials that will be used in contact with alveolar bone and apical periodontal connective tissues. In these cases, the implant site and the usage sites are sufficiently similar to compare the test results of the two sites.
Using In Vitro, Animal, and Usage Tests Together
For about 25 years, scientists, industry, and the government have recognized that the most accurate and cost-effective means to assess biocompatibility of a new material is a combination of in vitro, animal, and usage tests. Implicit in this philosophy is the concept that no single test will be adequate to completely characterize biocompatibility of a material. The ways in which these tests are used together, however, are controversial and have evolved over many years as knowledge has increased and new technologies were developed. This evolution can be expected to continue as materials are asked to perform more sophisticated functions for longer periods.
Early combination schemes proposed a pyramid testing protocol, in which all materials were tested at the bottom of the pyramid and materials were “weeded out” as the testing continued toward the top of the pyramid (Figure 6-6). Tests at the bottom of the pyramid were “unspecific toxicity” tests of any type (in vitro or animal) with conditions that did not necessarily reflect those of the material’s use. The next tier shows specific toxicity tests that presumably dealt with conditions more relevant to the use of the material. The final tier was a clinical trial of the material. Later, another pyramid scheme was proposed that divided tests into initial, secondary, and usage tests. The philosophy was similar to the first scheme, except that the types of tests were broadened to encompass biological reactions other than toxicity, such as immunogenicity and mutagenicity. The concept of a usage test in an animal was also added (versus a clinical trial in a human). There are several important features of these early schemes. First, only materials that “passed” the first tier of tests were graduated to the second tier, and only those that passed the second tier were graduated to the clinical trials.
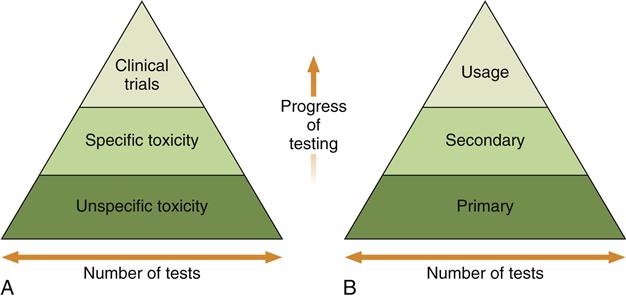
Testing begins at the bottom of the pyramid and works up. The number of tests needed decreases with the progress of testing because unacceptable materials are theoretically eliminated in the early testing stages. A, The earliest strategy, in which the testing strategy is focused on toxicity only. Unspecific toxicity refers to tests not necessarily related to the use of the material, whereas tests under specific toxicity are more relevant. Clinical trials are equivalent to usage tests in this scheme. B, The contemporary strategy used in most standards documents.
Presumably, then, this scheme funneled safer materials into the clinical trials area and eliminated unsafe materials. This strategy was welcomed because clinical trials are the most expensive and time-consuming aspect of biocompatibility testing. Second, any material that survived all three tiers of tests was deemed acceptable for clinical use. Third, each tier of the system put a great deal of weight on the tests used to accurately screen in or out a material. Although still used in principle today, the inability of in vitro and animal tests to unequivocally screen materials in or out has led to development of newer schemes in biocompatibility testing.
Two newer testing schemes have evolved in the past 5 years with regard to using combinations of biocompatibility tests to evaluate materials (Figure 6-7). Both of these newer schemes accommodate several important ideas. First, all tests (in vitro, animal, and usage) continue to be of value in assessing biocompatibility of a material during its development and even in its clinical service. For example, tests of inflammatory response in animals may be useful not only during the development of a material, but also if a problem is noted with the material after it has been on the market for a time. Second, these new schemes recognize the inability of current testing methods to accurately and absolutely screen in or out a material. Finally, they incorporate the philosophy that assessing the biocompatibility of a material is an ongoing process. Undoubtedly, we will see still newer strategies in the use of combinations of biocompatibility tests as the roles of materials change and the technologies for testing improve.
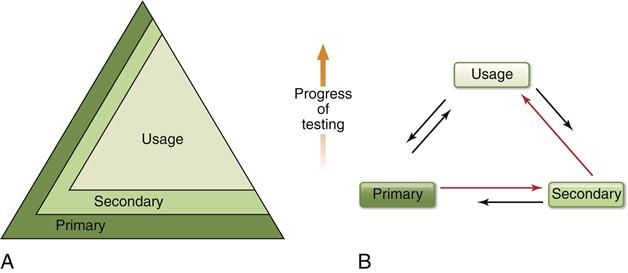
A, The pyramid scheme of Figure 6-6 is retained, but it is acknowledged that primary and secondary tests will play a continuing (but decreased) role as the progress of the testing continues. B, The usage test has the most stature and the most common progression of tests is from primary to secondary to usage, but the need to go through several iterations between testing types is acknowledged. Furthermore, the ongoing nature of biocompatibility is recognized by the need to use primary and secondary tests after clinical evaluation of a material. In this scheme the order of testing is ultimately determined as the testing and clinical use of the material continues to provide new data.
Standards That Regulate the Measurement of Biocompatibility
The first efforts of the American Dental Association (ADA) to establish guidelines for dental materials came in 1926 when scientists at the National Bureau of Standards (NBS), now the National Institute of Science and Technology (NIST), developed specifications for dental amalgam. Unfortunately, recommendations on materials and conditions for biological compatibility have not kept pace with the technological development of dental materials. Reasons for this are (1) the fast advance of cellular and molecular biology, (2) the variety of tests available for assessing biocompatibility of materials, and (3) the lack of standardization of these tests.
Standardization is a difficult and lengthy process, made more difficult by disagreement on the appropriateness and significance of particular tests. One of the early attempts to develop a uniform test for all materials was the study by Dixon and Rickert in 1933, in which the toxicity of most dental materials in use at that time was investigated by implanting the materials into pockets in subdermal tissue. Small, standard-sized pieces of gold, amalgam, gutta-percha, silicates, and copper amalgam were sterilized and placed in uniformly sized pockets within skeletal muscle tissue. Biopsy specimens were evaluated microscopically after 6 months. Other early attempts to standardize techniques were carried out by Mitchell (1959) on connective tissue and by Massler (1958) on tooth pulp. Not until the passage of the Medical Device Bill by Congress in 1976 was biological testing for all medical devices (including dental materials) given a high priority. In 1972 the ADA Council on Dental Materials, Instruments, and Equipment (now the Council on Scientific Affairs) approved specification No. 41 for Recommended Standard Practices for Biological Evaluation of Dental Materials. The committee that developed this document recognized the need for standardized methods of testing and for sequential testing of materials to reduce the number of compounds that would need to be tested clinically. In 1982, an addendum was made to this document, and it was further updated to its present form in 2005.
ANSI/ADA Specification 41
Three categories of tests are described in the 2005 ANSI/ADA (American National Standards Institute/American Dental Association) specification: initial, secondary, and usage tests. This document uses the testing scheme shown in Figure 6-6, B. The initial tests include in vitro assays for cytotoxicity, red blood cell membrane lysis (hemolysis), mutagenesis and carcinogenesis at the cellular level, and in vivo acute physiological distress and death at the level of the organism. Based on the results of these initial tests, promising materials are tested by one or more secondary tests in small animals (in vivo) for inflammatory or immunogenic potential (e.g., dermal irritation, subcutaneous and bony implantation, and hypersensitivity tests). Finally, materials that pass secondary tests and still hold potential are subjected to one or more in vivo usage tests, first in larger animals, often primates, and finally, with Food and Drug Administration approval, in humans. The ANSI/ADA specification No. 41, 1982 addendum, added two assays for mutagenesis—the Ames test and the Styles’ cell transformation test. The standard was most recently revised to conform to the ISO (International Organization for Standardization) standard below, and was released as ANSI/ADA specification No. 41, Recommended Standard Practices for Biological Evaluation of Dental Materials (2005).
ISO 10993
In the 1980s, international efforts were initiated by several organizations to develop international standards for biomedical materials and devices. Several multinational working groups, including scientists from ANSI and the ISO, were formed to develop standard ISO 10993, published in 1992. Revision of the dental components of this document resulted in ISO 7405:2008 “Preclinical evaluation of biocompatibility of medical devices used in dentistry—Test methods for dental materials.” This is the most recent ISO standard available for biocompatibility testing of dental materials.
The standard divides tests into initial and supplementary tests to assess the biological reaction to materials. Initial tests are tests for cytotoxicity, sensitization, and systemic toxicity. Some of these tests are done in vitro, others in animals in nonusage situations. Most of the supplementary tests, to assess things such as chronic toxicity, carcinogenicity, and biodegradation, are done in animal systems, many in usage situations. Significantly, although guidelines for the selection of tests are given in part 1 of the standard and are based on how long the material will be present; whether it will contact body surface only, blood, or bone; and whether the device communicates externally from the body, the ultimate selection of tests for a specific material is left up to the manufacturer, who must present and defend the testing results.
Biocompatibility of Dental Materials
Reactions of Pulp
Microleakage
There is evidence that restorative materials may not bond to enamel or dentin with sufficient strength to resist the forces of contraction during polymerization, wear, or thermal cycling. If a bond does not form, or debonding occurs, bacteria, food debris, or saliva may be drawn into the gap between the restoration and the tooth by capillary action. This effect has been termed microleakage. The importance of microleakage in pulpal irritation has been extensively studied. Several early studies reported that various dental restorative materials irritated pulp tissue in animal tests. However, several other studies hypothesized that the products of microleakage, not the restorative materials, caused the irritation. Subsequently, numerous studies showed that bacteria present under restorations and in dentinal tubules might be responsible for pulpal irritation. Other studies showed that bacteria or bacterial products such as lipopolysaccharides could cause pulp irritation within hours of being applied to dentin.
Finally, a classic animal study shed light on the roles of restorative materials and microleakage on pulpal irritation. Amalgam, composite, zinc phosphate cement, and silicate cement were used as restorative materials in class 5 cavity preparations in monkey teeth. The materials were placed directly on pulp tissues. Half of the restorations were surface-sealed with ZOE cement. Although some irritation was evident in all restorations at 7 days, after 21 days, the sealed restorations showed less pulpal irritation than those not sealed, presumably because microleakage had been eliminated. Only zinc phosphate cement elicited a long-term inflammatory response. Furthermore, the sealed teeth exhibited a much higher rate of dentin bridging under the material. Only amalgam seemed to prevent bridging. This study suggests that microleakage plays a significant role in pulpal irritation, but that the materials can also alter normal pulpal and dentinal repair.
Recently, the concept of nanoleakage has been put forward. Like microleakage, nanoleakage refers to the leakage of saliva, bacteria, or material components through the interface b/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


