The Etiology of Orthodontic Problems
Malocclusion is a developmental condition. In most instances, malocclusion and dentofacial deformity are caused, not by some pathologic process, but by moderate (occasionally severe) distortions of normal development. Occasionally, a single specific cause is apparent, for example, in mandibular deficiency secondary to a childhood fracture of the jaw or the characteristic malocclusion that accompanies some genetic syndromes. More often, these problems result from a complex interaction among multiple factors that influence growth and development, and it is impossible to describe a specific etiologic factor (Figure 5-1).
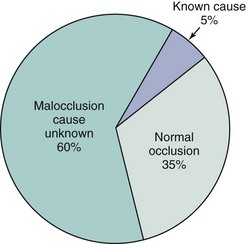
Specific Causes of Malocclusion
Disturbances in Embryologic Development
Defects in embryologic development usually result in death of the embryo. As many as 20% of early pregnancies terminate because of lethal embryologic defects, often so early that the mother is not even aware of conception. Although most defects in embryos are of genetic origin, effects from the environment also are important. Chemical and other agents capable of producing embryologic defects if given at the critical time are called teratogens. Most drugs do not interfere with normal development or, at high doses, kill the embryo without producing defects, and therefore are not teratogenic. Teratogens typically cause specific defects if present at low levels but if given in higher doses, do have lethal effects. Teratogens known to produce orthodontic problems are listed in Table 5-1.
TABLE 5-1
Teratogens Affecting Dentofacial Development
| Teratogens | Effect |
| Aminopterin | Anencephaly |
| Aspirin | Cleft lip and palate |
| Cigarette smoke (hypoxia) | Cleft lip and palate |
| Cytomegalovirus | Microcephaly, hydrocephaly, microphthalmia |
| Dilantin | Cleft lip and palate |
| Ethyl alcohol | Central midface deficiency |
| 6-Mercaptopurine | Cleft palate |
| 13-cis Retinoic acid (Accutane) | Similar to craniofacial microsomia and Treacher Collins syndrome |
| Rubella virus | Microphthalmia, cataracts, deafness |
| Thalidomide | Malformations similar to craniofacial microsomia, Treacher Collins syndrome |
| Toxoplasma | Microcephaly, hydrocephaly, microphthalmia |
| X-radiation | Microcephaly |
| Valium | Similar to craniofacial microsomia and Treacher Collins syndrome |
| Vitamin D excess | Premature suture closure |
There are five principal stages in craniofacial development (Table 5-2), and effects on the developing face and jaws can arise during each stage:
TABLE 5-2
Stages of Embryonic Craniofacial Development
| Stage | Time in humans (postfertilization) | Related syndromes |
| Germ layer formation and initial organization of structures | Day 17 | Fetal alcohol syndrome (FAS) |
| Neural tube formation | Days 18-23 | Anencephaly |
| Origin, migration, and interaction of cell populations | Days 19-28 | Craniofacial microsomia Mandibulofacial dysostosis (Treacher Collins syndrome) Limb abnormalities |
| Formation of organ systems Primary palate Secondary palate |
Days 28-38 Days 42-55 |
Cleft lip and/or palate, other facial clefts Cleft palate |
| Final differentiation of tissues | Day 50-birth | Achondroplasia Synostosis syndromes (e.g., Crouzon’s, Apert’s) |
1. Germ layer formation and initial organization of craniofacial structures
2. Neural tube formation and initial formation of the oropharynx
3. Origins, migrations, and interactions of cell populations, especially neural crest cells
4. Formation of organ systems, especially the pharyngeal arches and the primary and secondary palates
5. Final differentiation of tissues (skeletal, muscular, and nervous elements)
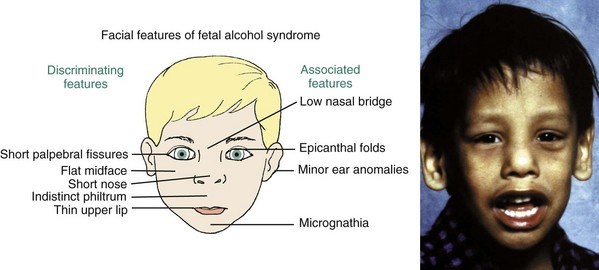
The best example of a problem that can be traced to the very early first and second stages is the characteristic facies of fetal alcohol syndrome (FAS; Figure 5-2). This is due to deficiencies of midline tissue of the neural plate very early in embryonic development caused by exposure to very high levels of ethanol. Although such blood levels can be reached only in extreme intoxication in chronic alcoholics, the resulting facial deformity and developmental delay occur frequently enough to be implicated in many cases of midface deficiency.1 In these unfortunate children, the delay in dental development matches the skeletal delay.2
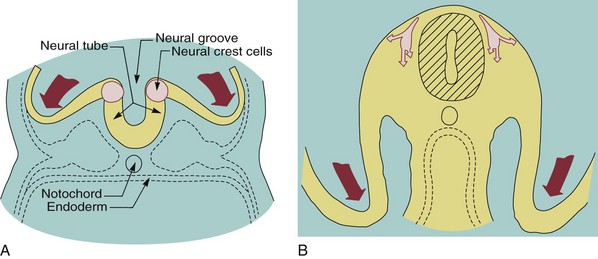
Many of the problems that result in craniofacial anomalies arise in the third stage of development and are related to neural crest cell origin and migration. Since most structures of the face are ultimately derived from migrating neural crest cells (Figure 5-3), it is not surprising that interferences with this migration produce facial deformities. At the completion of the migration of the neural crest cells in the fourth week of human embryonic life, they form practically all of the loose mesenchymal tissue in the facial region that lies between the surface ectoderm and the underlying forebrain and eye and most of the mesenchyme in the mandibular arch. Most of the neural crest cells in the facial area later differentiate into skeletal and connective tissues, including the bones of the jaw and the teeth.
The importance of neural crest migration and the possibility of drug-induced impairment of the migration have been demonstrated clearly by unfortunate experience. In the 1960s and 1970s, exposure to thalidomide caused major congenital defects, including facial anomalies in thousands of children. In the 1980s, severe facial malformations related to the anti-acne drug isotretinoin (Accutane) were reported. The similarities in the defects make it likely that both these drugs affect neural crest cells. Retinoic acid plays a crucial role in the ontogenesis of the midface, and recent work suggests that loss of retinoic acid receptor genes affects postmigratory activity of crest cells, clarifying the timing of Accutane effects.3 The danger with isotretinoin is that it affects a developing embryo before the mother knows she is pregnant.
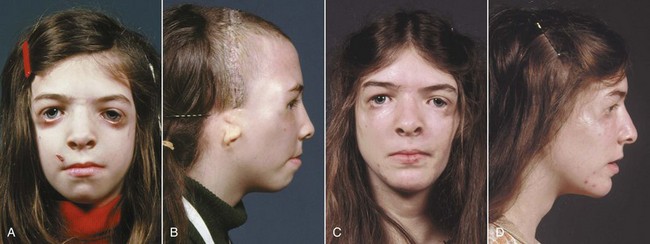
Altered development of cells derived from the neural crest also has been implicated in Treacher Collins syndrome (Figure 5-4), which is characterized by a generalized lack of mesenchymal tissue and now known to be due (at least in some instances) to mutations in a specific gene (TCOF1) that lead to loss of a specific exon.4
Craniofacial microsomia (formerly called hemifacial microsomia) is characterized by a lack of development in lateral facial areas. Typically, the external ear is deformed and both the ramus of the mandible and associated soft tissues (muscle, fascia) are deficient or missing (Figure 5-5). Although facial asymmetry is always seen (thus the former name), cranial as well as facial structures are affected. The cause is loss of neural crest cells (for an unknown reason) during migration. Neural crest cells with the longest migration path, those taking a circuitous route to the lateral and lower areas of the face, are most affected, whereas those going to the central face tend to complete their migratory movement, so midline facial defects, including clefts, rarely are part of the syndrome.5

The most common congenital defect involving the face and jaws, second only to clubfoot in the entire spectrum of congenital deformities, is clefting of the lip, palate, or (less commonly) other facial structures. Clefts arise during the fourth developmental stage. Exactly where they appear is determined by the locations at which fusion of the various facial processes failed to occur (Figures 5-6 and 5-7), and this in turn is influenced by the time in embryologic life when some interference with development occurred.
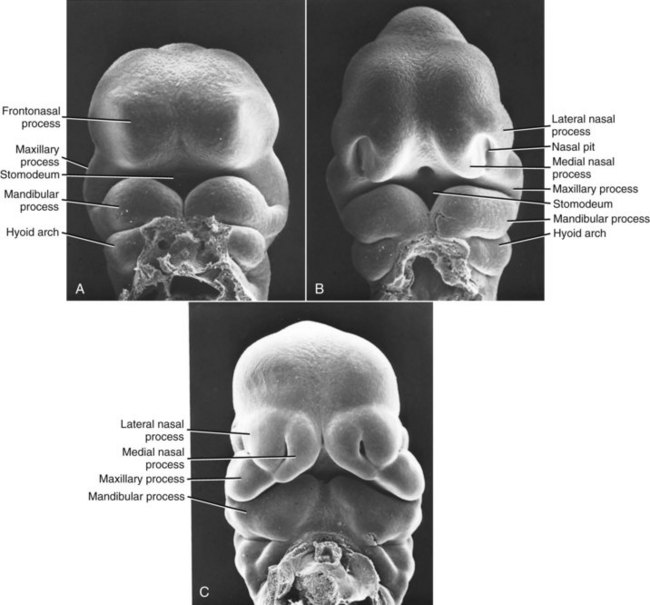
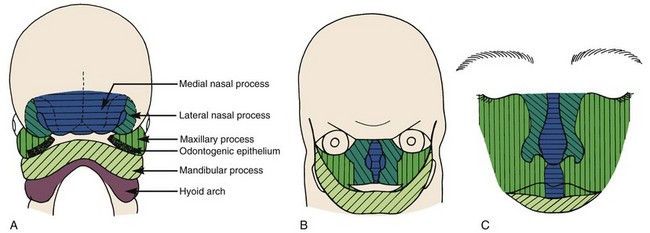
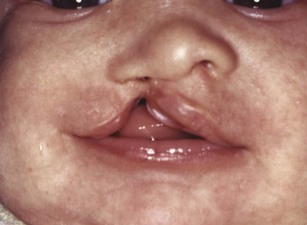
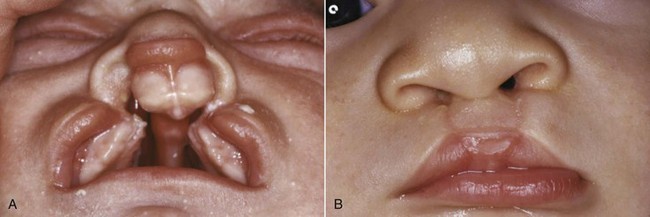
Clefting of the lip occurs because of a failure of fusion between the median and lateral nasal processes and the maxillary prominence, which normally occurs in humans during the sixth week of development. At least theoretically, a midline cleft of the upper lip could develop because of a split within the median nasal process, but this almost never occurs. Instead, clefts of the lip occur lateral to the midline on either or both sides (Figure 5-8). Since the fusion of these processes during primary palate formation creates not only the lip but the area of the alveolar ridge containing the central and lateral incisors, it is likely that a notch in the alveolar process will accompany a cleft lip even if there is no cleft of the secondary palate.
Closure of the secondary palate by elevation of the palatal shelves (Figures 5-9 and 5-10) follows that of the primary palate by nearly 2 weeks, which means that an interference with lip closure that still is present can also affect the palate. About 60% of individuals with a cleft lip also have a palatal cleft (Figure 5-11). An isolated cleft of the secondary palate is the result of a problem that arose after lip closure was completed. Incomplete fusion of the secondary palate produces a notch in its posterior extent (sometimes only a bifid uvula). This indicates a very late-appearing interference with fusion.
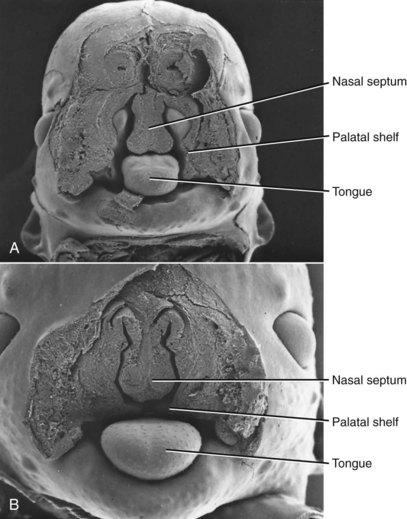
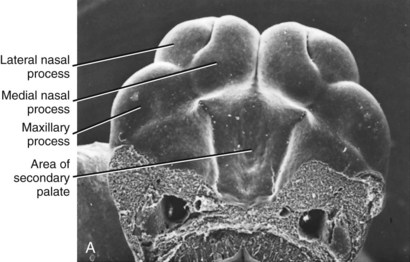
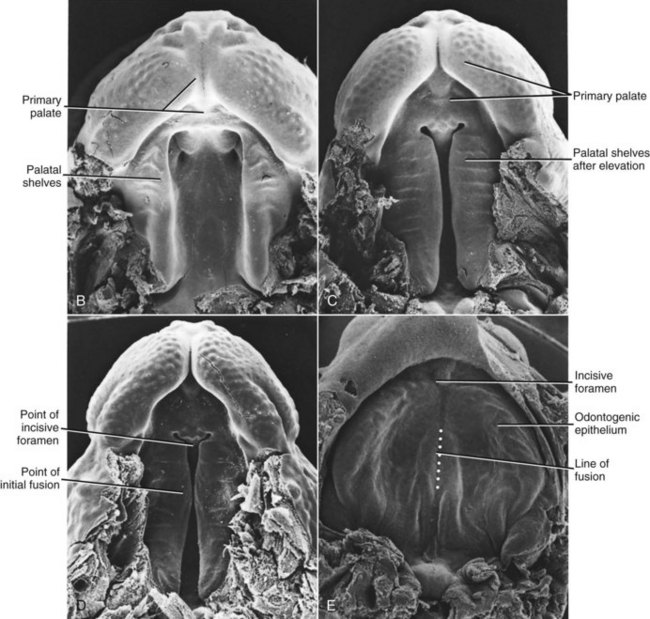
FIGURE 5-10 Scanning electron micrographs of the stages in palate closure (mouse embryos sectioned so that the lower jaw has been removed), analogous to the same stages in human embryos. A, At the completion of primary palate formation. B, Before elevation of the palatal shelves, equivalent to Figure 3-8, A. C, Shelves during elevation. D, Initial fusion of the shelves at a point about one third of the way back along their length. E, Secondary palate immediately after fusion. (Courtesy Dr. K. Sulik.)
The width of the mouth is determined by fusion of the maxillary and mandibular processes at their lateral extent, so a failure of fusion in this area could produce an exceptionally wide mouth, or macrostomia. Failure of fusion between the maxillary and lateral processes could produce an obliquely directed cleft of the face. Other patterns of facial clefts are possible, based on the details of fusion, and were classified by Tessier.6 Fortunately, these conditions are rare.
Morphogenetic movements of the tissues are a prominent part of the fourth stage of facial development. As these have become better understood, the way in which clefts of the lip and palate develop has been clarified. For example, it is known now that cigarette smoking by the mother is an etiologic factor in the development of cleft lip and palate,7 and even passive smoke increases the risk of cleft palate.8 An important initial step in development of the primary palate is a forward movement of the lateral nasal process, which positions it so that contact with the median nasal process is possible. The hypoxia associated with smoking probably interferes with this movement.
Another major group of craniofacial malformations arise considerably later than the ones discussed so far, during the final stage of facial development and in the early fetal rather than the embryologic period of prenatal life. These are the craniosynostosis syndromes, which result from early closure of the sutures between the cranial and facial bones. In fetal life, normal cranial and facial development depends on growth adjustments at the sutures in response to growth of the brain and facial soft tissues. Early closure of a suture, called synostosis, leads to characteristic distortions, depending on the location of the early fusion.9
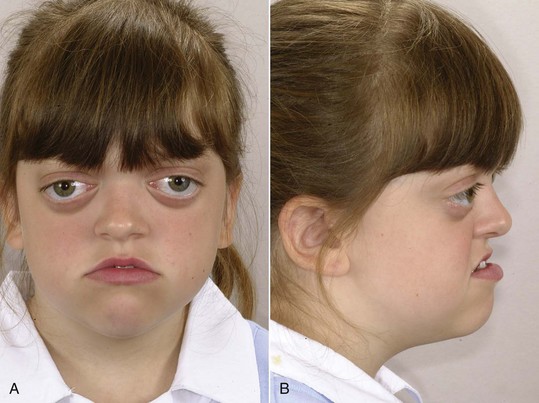
Crouzon’s syndrome is the most frequently occurring member of this group. It is characterized by underdevelopment of the midface and eyes that seem to bulge from their sockets (Figure 5-12). This syndrome arises because of prenatal fusion of the superior and posterior sutures of the maxilla along the wall of the orbit. The premature fusion frequently extends posteriorly into the cranium, producing distortions of the cranial vault as well. If fusion in the orbital area prevents the maxilla from translating downward and forward, the result is severe underdevelopment of the middle third of the face. The characteristic protrusion of the eyes is largely an illusion—the eyes appear to bulge outward because the area beneath them is underdeveloped. There may be a component of true extrusion of the eyes, however, because when cranial sutures become synostosed, intracranial pressure increases.
Growth Disturbances in the Fetal and Perinatal Period
Fetal Molding and Birth Injuries
Intrauterine Molding: Pressure against the developing face prenatally can lead to distortion of rapidly growing areas. Strictly speaking, this is not a birth injury, but because the effects are noted at birth, it is considered in that category. On rare occasions, an arm is pressed across the face in utero, resulting in severe maxillary deficiency at birth (Figure 5-13). Occasionally, a fetus’ head is flexed tightly against the chest in utero, preventing the mandible from growing forward normally. This is related to a decreased volume of amniotic fluid, which can occur for any of several reasons. The result is an extremely small mandible at birth, usually accompanied by a cleft palate because the restriction on displacement of the mandible forces the tongue upward and prevents normal closure of the palatal shelves.
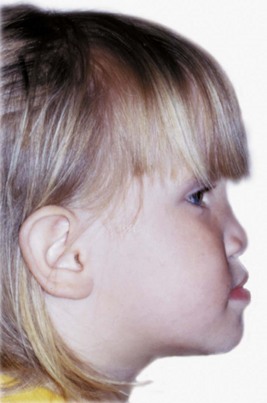
Because the pressure against the face that caused the growth problem would not be present after birth, there is the possibility of normal growth thereafter and perhaps eventually a complete recovery. Some children with Pierre Robin sequence at birth do have favorable mandibular growth in childhood, but a smaller than normal mandible typically persists (Figure 5-14), and a recent study found no catch-up growth during adolescence.10 It has been estimated that about one-third of the Pierre Robin patients have a defect in cartilage formation and can be said to have Stickler syndrome. Not surprisingly, this group has limited growth potential. Catch-up growth is most likely when the original problem was mechanical growth restriction that no longer existed after birth.
Birth Trauma to the Mandible: Many facial deformity patterns now known to result from other causes once were blamed on injuries during birth. Many parents, despite explanations from their doctors, will refer to their child’s facial deformity as being caused by a birth injury even if a congenital syndrome is evident. No matter what the parents say later, a recognizable syndrome obviously did not arise because of birth trauma.
Progressive Deformities in Childhood
Childhood Fractures of the Jaw
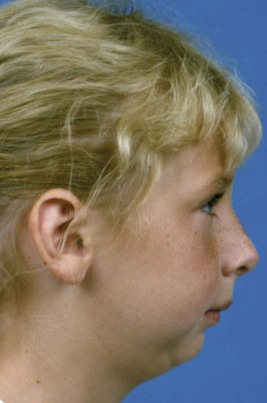
When a problem does arise following condylar fracture, it usually is asymmetric growth deficiency, with the injured side (or, in bilateral fractures, the more severely injured side) lagging behind (Figure 5-15). After an injury, if there is enough scarring around the TM joint to restrict translation of the condyle, so that the mandible cannot be pulled forward as much as the rest of the growing face, subsequent growth will be restricted.
This concept is highly relevant to the management of condylar fractures in children. It suggests, and clinical experience confirms, that there would be little if any advantage from surgical open reduction of a condylar fracture in a child. The additional scarring produced by surgery could make things worse. The best therapy therefore is conservative management at the time of injury and early mobilization of the jaw to minimize any restriction on movement. If deficient growth is observed, however, early treatment is needed (see Chapter 12).
Although an old condylar fracture is the most likely cause of asymmetric mandibular deficiency in a child, other destructive processes that involve the TM joint, such as rheumatoid arthritis (Figure 5-16), or a congenital absence of tissue as in craniofacial microsomia also can produce this problem.
Muscle Dysfunction
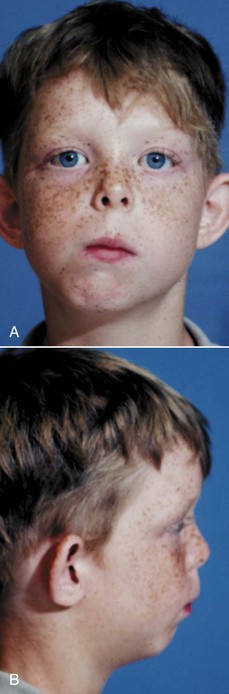
The facial muscles can affect jaw growth in two ways. First, the formation of bone at the point of muscle attachments depends on the activity of the muscle; second, the musculature is an important part of the total soft tissue matrix whose growth normally carries the jaws downward and forward. Loss of part of the musculature is most likely to result from damage to the motor nerve (muscle atrophies when its motor nerve supply is lost). The result would be underdevelopment of that part of the face, with a deficiency of both soft and hard tissues (Figure 5-17).
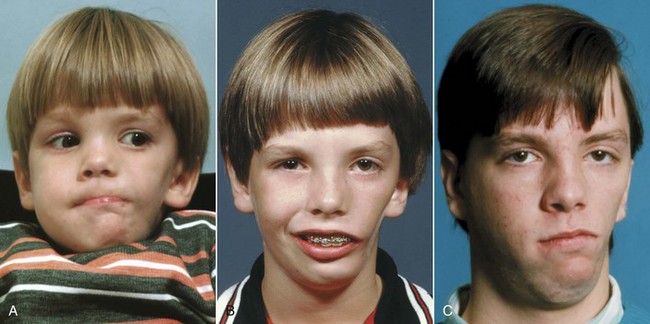
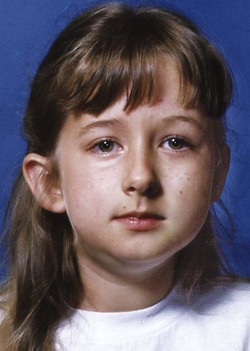
Excessive muscle contraction can restrict growth in much the same way as scarring after an injury. This effect is seen most clearly in torticollis, a twisting of the head caused by excessive tonic contraction of the neck muscles on one side (primarily the sternocleidomastoid) (Figure 5-18). The result is a facial asymmetry because of growth restriction on the affected side, which can be quite severe unless the contracted neck muscles are surgically detached at an early age.11 Conversely, a major decrease in tonic muscle activity (as in muscular dystrophy, some forms of cerebral palsy, and various muscle weakness syndromes) allows the mandible to drop downward away from the rest of the facial skeleton. The result is increased anterior face height, distortion of facial proportions and mandibular form, excessive eruption of the posterior teeth, narrowing of the maxillary arch, and anterior open bite (Figure 5-19).12
Disturbances Arising in Adolescence or Early Adult Life
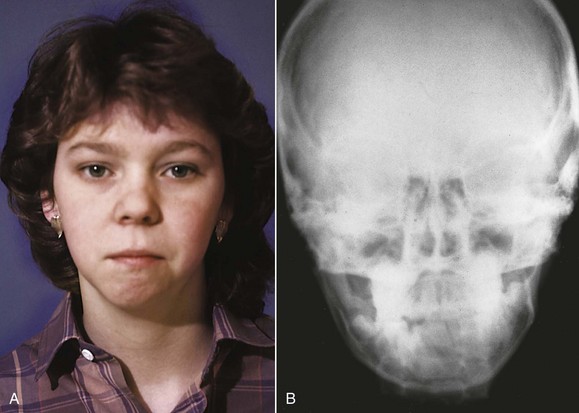
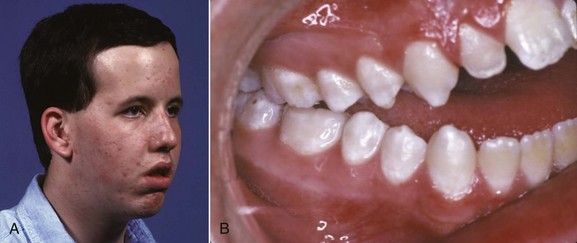
Occasionally, unilateral excessive growth of the mandible occurs in individuals who seem metabolically normal. Why this occurs is entirely unknown. It is most likely in girls between the ages of 15 and 20 but may occur as early as age 10 or as late as the early 30s in either sex. The condition formerly was called condylar hyperplasia, and proliferation of the condylar cartilage is a prominent aspect; however, because the body of the mandible also is affected (Figure 5-20), hemimandibular hypertrophy now is considered a more accurate descriptive term.13 The excessive growth may stop spontaneously, but in severe cases removal of the affected condyle and reconstruction of the area are necessary.
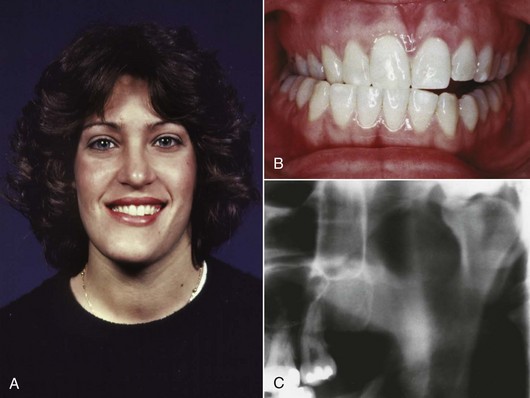
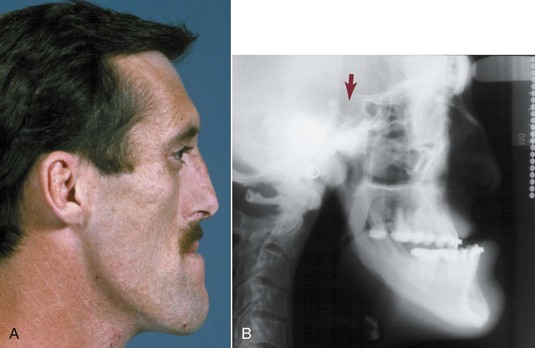
In acromegaly, which is caused by an anterior pituitary tumor that secretes excessive amounts of growth hormone, excessive growth of the mandible may occur, creating a skeletal Class III malocclusion in adult life (Figure 5-21). Often (but not always—sometimes the mandible is unaffected while hands and/or feet grow), mandibular growth accelerates again to the levels seen in the adolescent growth spurt, years after adolescent growth was completed.14 The condylar cartilage proliferates, but it is difficult to be sure whether this is the cause of the mandibular growth or merely accompanies it. Although the excessive growth stops when the tumor is removed or irradiated, the skeletal deformity persists and orthognathic surgery to reposition the mandible is likely to be necessary (see Chapter 19).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


