chapter 4
The Processing of Pain at the Brainstem Level
Now that the normal anatomy and physiology of the peripheral and central nervous systems (CNS) have been described, a more thorough description of the manner by which pain is initiated and processed can be reviewed. An understanding of how noxious impulses are credited in the periphery and are processed by the CNS is essential for the clinician who wishes to manage pain disorders.
The Initiation of Nociception in the Peripheral Tissues
When tissue is injured, the damaged cells release potassium (K+), which initiates the synthesis and release of bradykinin, a nine-amino acid peptide, from large plasma proteins. Bradykinin is a very potent pain-producing substance. Tissue injury also causes the breakdown of arachidonic acid into prostaglandins by the enzymatic action of cyclooxygenase (COX). Arachidonic acid is also converted to leukotrienes by the enzymatic action of 5-lipoxygenase (see chapter 2). The presence of these substances activates the A-delta and C fibers in the immediate region of the injury. The initial activation of the A-delta fiber produces a quick volley of afferent nociceptive input into the CNS, signaling a sharp, acute pain. Prostaglandins also sensitize the nociceptors to bradykinin, substance P (SP), and other algesic substances, which now begin to discharge, causing sensitization of the slower conducting C fibers. The pain experience now becomes more of a dull, aching, sometimes burning sensation.
Although the tissue injury may only originally affect one portion of the primary afferent neuron, a series of events often takes place that leads to an expansion of the involved area. This occurs through the antidromic release of algesic substances. When an afferent nerve pathway is discussed, the term orthodromic means that the transmission of impulses is passing from the periphery into the CNS. The term antidromic means that the pathway of transmission is actually reversed, so that an afferent neuron is functioning in reverse, causing changes to occur in the periphery. This is precisely what occurs at the branches of a primary afferent neuron when a single branch is injured. With injury, SP and calcitonin gene-related peptide (CGRP) are antidromically released in the other peripheral branches of the same afferent neuron (Fig 4-1). SP then causes mast cells in the area to release histamine and the platelets to release serotonin (5-hydoxy-tryptamine [5HT]). Histamine, CGRP, and 5HT mediate swelling, redness, and heat and increase peripheral sensitivity to further stimulation.1 This leads to hyperalgesia (increased sensitivity to stimulation-evoked pain). As the level of histamine and 5HT increases in the extracellular space, there is more sensitization of neighboring nociceptors, leading to a spreading of the hyperalgesic area.2 Additionally, SP causes the blood vessels in the area to release more bradykinin into the site of injury, leading to further sensitization. This spreading of the painful area beyond the site of the actual injury is called primary hyperalgesia.
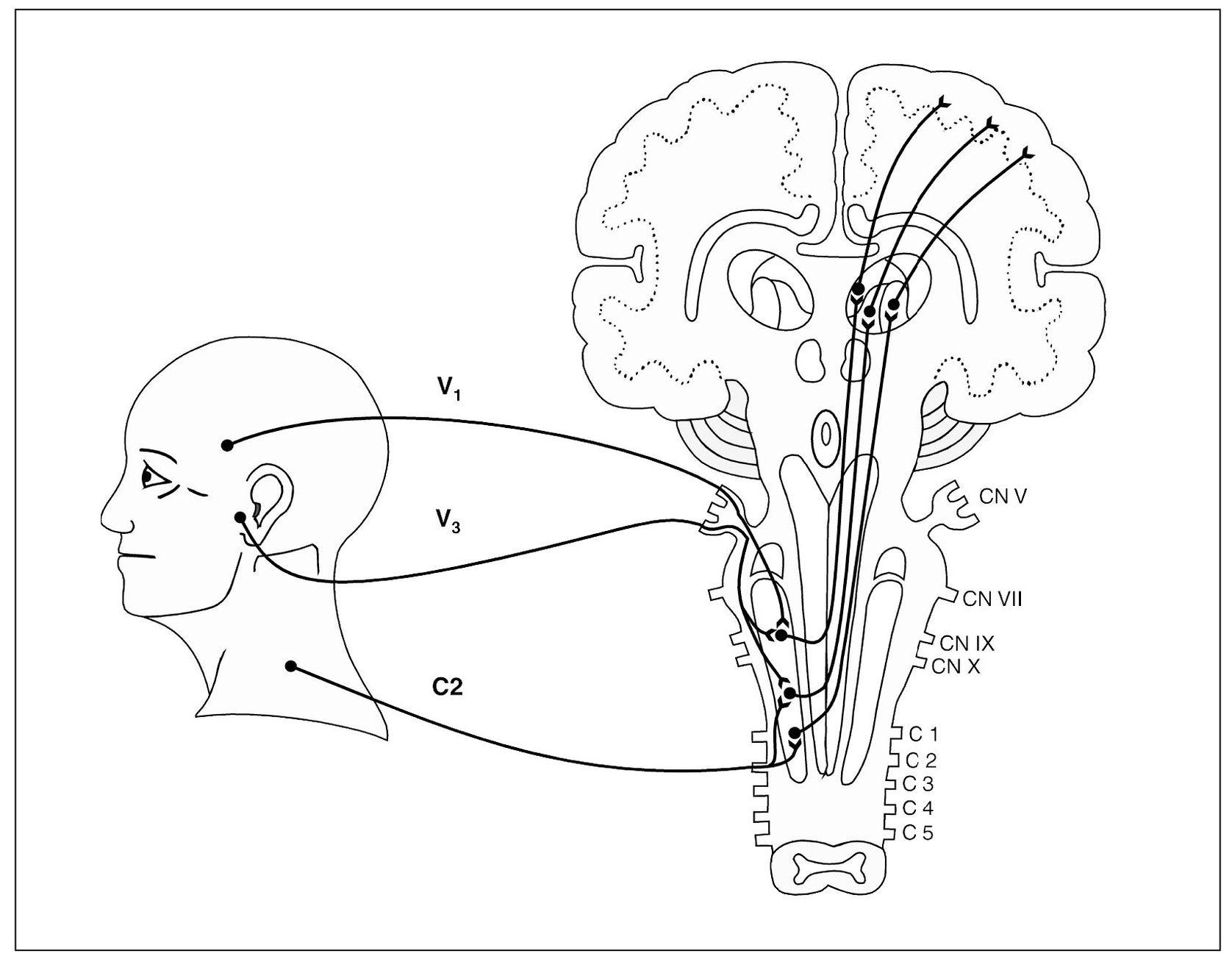
Fig 4-1 A simplified graphic depiction of convergence of the primary neurons in the nucleus caudalis region of the trigeminal spinal tract nucleus. The neuron representing the ophthalmic division (V1) and the neuron representing the mandibular division (V3) of the trigeminal nerve converge on the same second-order neuron. The trigeminal spinal tract nucleus extends caudally to the region of the entrance zone of upper cervical nerves. The second cervical nerve (C2) enters this region of the brainstem and converges on both a cervical-input, second-order neuron as well as a second-order neuron that is also receiving trigeminal input.
The Central Processing of Nociception
Convergence
It has been known for many years that there are more primary afferent neurons entering the CNS than there are second-order neurons to carry the impulses on to the higher centers.3 It therefore follows that several primary sensory neurons must synapse with a single second-order neuron. The synapsing of several primary afferent neurons with one second-order neuron is known as convergence. In the same sense, a single primary afferent neuron can also enter the CNS and synapse with more than one second-order neuron. This condition is called divergence. Activity at the synapse may be a cumulative effect, called summation. When several afferent neurons simultaneously stimulate the same second-order neuron, the summation is called spatial. Summation can also occur when a single neuron stimulates a second-order neuron in rapid succession. This is known as temporal summation. Intensification of response is known as facilitation; suppression of response is called inhibition.
Convergence has been well documented in the trigeminal brainstem sensory nuclear complex. The subnucleus oralis and interpolaris receive extensive convergence of orofacial and muscle afferent inputs4,5,6,7,8,9,10,11,12,13 (Fig 4-2). The same convergence has been shown in the subnucleus caudalis. In fact, in one study14 of the cat brain, almost all of the neurons with input from the temporo-mandibular joint (TMJ) received additional afferent input, predominantly from facial skin or intraoral sites. In this same study, 74% of the neurons tested in the subnucleus caudalis showed convergence of tooth pulp and/or hypoglossal nerve afferent inputs. In another study,15 afferent input from the TMJ and masseter muscle converged on the same second-order neuron in 80% of the nerves that were tested in the subnucleus caudalis. It appears that afferent inputs from deep structures converge to a greater degree than do afferents from cutaneous structures.16,17 Perhaps this is why pain from deep structures is felt to be more diffuse and less localized than the more localized pain that is felt from cutaneous structures. In fact, deep pains are often very difficult for the patient to locate. As will be discussed in the next section, convergence of nociceptive input can lead to a confusing diagnostic clinical presentation.
Site of Pain Versus Source of Pain
For the clinician to successfully evaluate pain disorders, he or she must appreciate the difference between the site and the source of pain. Although these words are often used interchangeably, they have significantly different meanings. The site of pain is the location that the patient feels the pain. The site of pain is easily located by merely asking the patient to point out the region of the body that is painful. The source of pain is that area of the body from which the pain actually originates. When the site and the source of pain are in the same location, it is called primary pain. Primary pains are very common and likely to be the only type of pain familiar to the patient. When one cuts a finger, the area of tissue damage is also the location of the pain. This is an example of primary pain, since the site of pain (where it hurts) and the source of pain (where it originates) are in the same location. This type of pain makes sense to the patient and the clinician, and therefore therapy is obvious.
There are pains, however, in which the site of the pain is not in the same location as the source of the pain. This type of pain is called heterotopic pain. Heterotopic pains can be quite confusing to both the patient and the clinician. A commonly known heterotopic pain is cardiac pain. When an individual experiences myocardial ischemia, the site of pain is frequently felt in the mandible, in the shoulder, or radiating down the left arm. The source of pain is the myocardial tissues, yet the site of pain is distant from the site. Heterotopic pains pose a significant problem for the clinician. A basic cardinal rule in therapy is that treatment must be directed toward the source of pain, not the site of pain. Treatments directed toward the site of pain will have little to no effect on the pain. The clinician must therefore be able to first recognize heterotopic pains and then differentiate the site from the source of pain. This is basic to orofacial pain therapy, since heterotopic pains are extremely common in the head and neck regions. Other common examples of heterotopic pain in the head and neck are frontal and temporal headaches from the neck, and ear and temporomandibular joint pain produced from the sternocleidomastoid muscle. Sooner or later, every dental practitioner is confronted with a patient who complains of tooth pain but may not present any objective findings. As will be discussed later, many toothaches do not arise from the teeth at all.
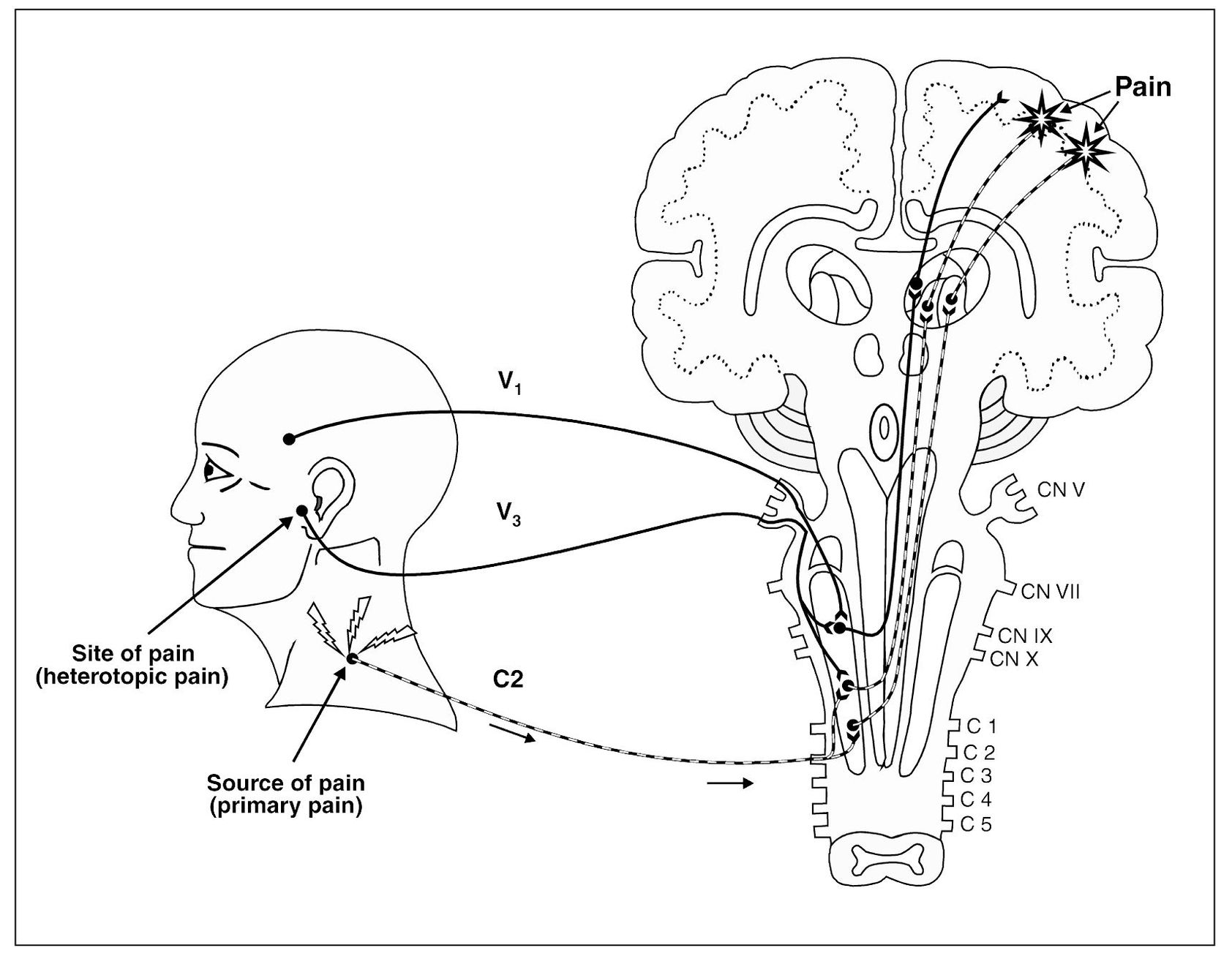
Fig 4-2 Injury to the trapezius muscle results in tissue damage. Nociception arising in this cervical region is transmitted to the second-order neuron and relayed on to the higher centers for interruption. As this input becomes protracted, note that the adjacent converging neuron is also centrally excited, which relays additional nociception on to the higher centers. The sensory cortex now perceives two locations of pain. One area is the trapezius region, which represents a true source of nociception (primary pain). The second area of perceived pain is felt in the temporomandibular joint area, which is only a site of pain, not a source of pain. This pain is heterotopic (referred).
Each clinician must be prepared to accept the fact that pain, especially from deep sources, is frequently felt in structures that are normal. The clinician must therefore guard against extending treatment to such structures in an attempt to control pain until an accurate diagnosis has been made.
Types of Heterotopic Pains
The term heterotopic pain refers to any pain that is felt in an area other than its true source. Although the terms referred and heterotopic are frequently used interchangeably, a more restricted usage of the term referred is advocated. There are three general types of heterotopic pain: (1) central pain, (2) projected pain, and (3) referred pain.
Projected pain
Projected pain is felt in the peripheral distribution of the same nerve that mediates the primary nociceptive input. Pain resulting from noxious stimulation of a sensory root or a major nerve trunk is felt in the exact anatomic distribution of that nerve. An example is the radicular pain of posterior root compression. It has been assumed that compression due to herniation of an intervertebral disc produces prolonged firing in the injured sensory fibers. It should be noted, however, that acute peripheral nerve compression is usually painless and rarely lasts more than a few seconds. It is likely, therefore, that radicular pain is felt in the peripheral structures through activation of deeper interneurons.18 The mechanism, however, seems to be different from that of central excitation, in that projected pain follows a more precise anatomic pattern relative to the peripheral distribution, while central excitation pain is felt in a reference zone that is only segmentally related to the primary pain source. Projected sensory nerve pain is primarily neurogenous and follows dermatome mapping faithfully. Examples of projected pain include paroxysmal neuralgia, peripheral neuritis, herpes zoster, and postherpetic neuralgia.
It has long been known that noxious stimulation of a motor root or major motor nerve also induces pain, but of a different type that is felt in different areas. Rather than the bright, burning, neuropathic pain felt in the dermatome distribution, motor nerve pain is sensed as dull, deep somatic pain diffusely located in the muscles innervated by that nerve.19 Such pain is now explicable on the basis of afferent neurons that are present in the motor nerves. Undoubtedly, such pain should be classified as projected pain, even though it displays a deep somatic rather than neuropathic quality and follows a motor nerve route rather than a dermatome field. Although the neural mechanisms have not been identified, it is likely that interneurons are involved in a manner similar to that for projected sensory nerve pain. It likewise differs from the heterotopic pain of referred pain.
Projected pains may be accompanied by areas of secondary hyperalgesia that are hypersensitive to stimulation without an appreciable reduction in the local pain threshold. Whatever the mechanism responsible for this phenomenon, it is likely similar to that of secondary hyperalgesia of central hyperexcitability, as described in the next section.
Referred pain
Referred pain is a spontaneous heterotopic pain that is felt in an area innervated by a different nerve than the one that mediates the primary pain. Since it is spontaneous, referred pain occurs without provocation at the site of pain and is wholly dependent upon the original source of pain. The reason for this dependence is because the original source of nociceptive input produces a sensitization of interneurons that is responsible for this type of heterotopic pain.20,21,22,23,24,25 Since the sensitization of interneurons (also called central excitatory effects) is very important in understanding heterotopic pains, it is worthwhile to discuss this phenomenon at this time.
Central Sensitization
The diagnostic differentiation between true primary pain and symptoms that occur as secondary effects of that pain is essential. Such manifestations are usually referred to as central excitatory effects, on the assumption that they result from hyperexcitability of CNS interneurons.
The neurologic mechanisms involved in the secondary effects of pain are not fully understood. Some researchers think that reflex activity is involved.26 There seems to be little doubt that convergence of afferent impulses among CNS interneurons takes place.9,27,28 However, the factors that control the mechanisms involved have not been elucidated. Sessle et al29 have shown that, while primary trigeminal neurons normally respond only to stimuli located within their respective receptive fields, at least half of the second-order nociceptive neurons (ie, wide dynamic range and nociceptive-specific neurons) are activated by electric stimulation applied outside the normal receptive fields of the corresponding primary neurons. The neurons thus activated are presumed to be those that respond to input from deep structures of the mouth and face. This is taken as evidence that the reference of pain occurs in conjunction with deep pain input, rather than a superficial one.30 It suggests that under certain conditions, trigeminal nociceptive interneurons are subject to subliminal stimulation from structures other than those located in the normal receptive fields. Such evidence supports the convergence theory of pain reference. Gross31 reported that referred pain due to visceral disease followed known dermatomes, but referred pain from deep somatic structures did not.
Studies32 demonstrate that the function of a second-order neuron can actually change according to the type and intensity of input that the neuron receives. As discussed in the previous chapter, when a second-order neuron receives a constant barrage of nociceptive input, the cell itself activates cellular genes that change its function to these demands. This induction of early gene expression causes the release of proto-oncogenes called c-fos and c-jun.33 The release of these substances by the cell alters messenger RNA (mRNA), which in turn can change the type and number of receptors that are formed on the cell membrane. As the number and type of receptors change, so also does the cell’s function. These changes can result in a lowering of the firing threshold of the cell, thus sensitizing it to future stimulation. This is known as sensitization, and since the neurons being discussed here are in the CNS, this is referred to as central sensitization. The fact that a neuron can react and change function according to its input has only recently been appreciated. This process is called neuroplasticity and is nature’s way to adapt to environmental demands. Changes such as these can alter the reactivity of the neuron for minutes, days, months, or even years.
Some of the receptors that become involved when sensitization is prolonged are the N-methyl-D-aspartate (NMDA) receptors. 23,34,35,36,37 Stimulation of these receptors by excitatory amino acids, such as aspartate and glutamate, further increases the sensitization of the neurons. This increased sensitization can alter neural impulses as they are processed on the way to the higher centers. This is referred to as central excitatory effects. When this occurs, even normally nonnoxious input may be misinterpreted at the higher centers.
Clinically, sensitization and central excitatory effects are induced by more or less continuous barrages of noxious sensation emanating from deep somatic structures. If the input is not continuous, these secondary effects are less likely to occur. Increased intensity and duration of the deep pain input enhances the secondary effects. Initially, these changes in neural sensitization are usual and reversible. With chronicity, however, changes may occur that alter the neural processing more permanently. Permanent alterations can lead to chronic neuropathic pain, as will be discussed in chapter 17. The symptoms of central sensitization complicate continuous deep somatic pains, since they increase with the intensity and duration of the primary pain. Such symptoms can occur in otherwise normal structures, and therefore treatment can be misdirected.
The central excitatory effects produced by deep pain input may involve afferent (sensory) neurons, efferent (motor) neurons, and/or autonomic neurons. Each of these effects will now be discussed.
Afferent (sensory) neuron effects
When central excitatory effects are produced in a sensory neuron, the most common clinical finding is pain. The type of pain complaint is either referred pain or secondary hyperalgesia.
Referred pain
Referred pain due to central sensitization is initially wholly spontaneous as far as the site of pain is concerned. It is not accentuated by provocation of the site where the pain is felt; it is accentuated only by manipulation of the primary pain source (see Fig 4-2). It is dependent on continuation of the primary initiating pain and ceases immediately if the primary pain is arrested or interrupted. Anesthesia of the structure where the referred pain is felt does not arrest the pain.38 Only by anesthesia of the neural pathway that mediates the primary pain can the referred pain be arrested.39
It should be noted that although the primary initiating pain is of the deep somatic type, the secondary referred pain may be felt in either deep or superficial structures. This may present a bizarre clinical picture. When reference is felt superficially, it must be differentiated from superficial somatic pain and from projected neuropathic pain. When it is felt deeply, it must be differentiated from other deep somatic pains.
Referred pain does not occur haphazardly, but in fact follows three clinical rules. Referred pain most frequently occurs within a single nerve root, passing from one branch to another. The trigeminal nerve is a good illustration, since it has three major branches. For example, when a mandibular molar presents with a source of pain (eg, caries) it is not uncommon to have the patient report that a maxillary molar is also painful. In this case, the mandibular branch of the trigeminal nerve is referring pain to the maxillary branch of the same nerve.
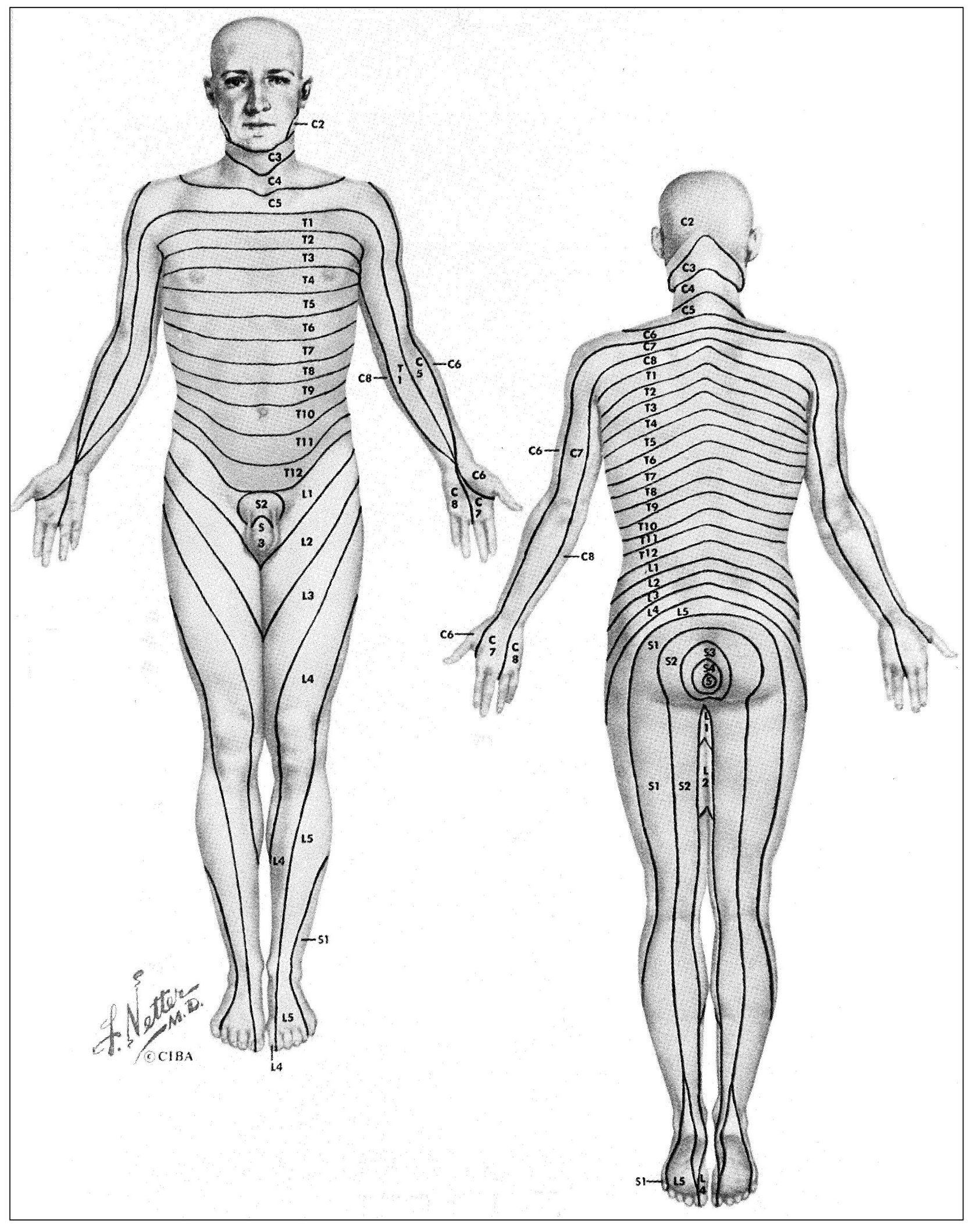
Fig 4-3 Dermal segmentation as represented by Netter and based essentially on the work of Keegan. (From Netter FH. The CIBA Collection of Medical Illustrations. Indianapolis: Curtis, 1953. Used with permission.)
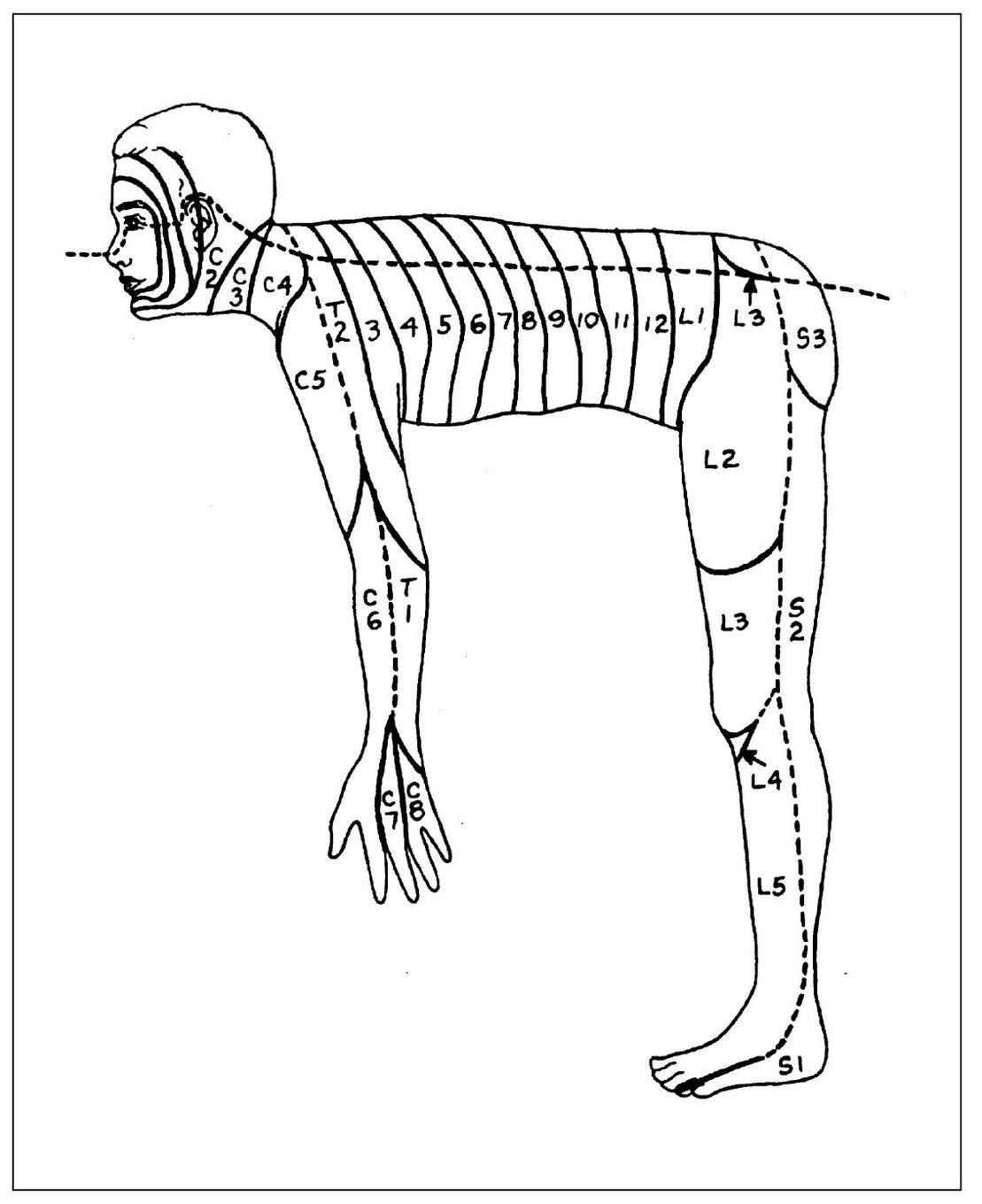
Fig 4-4 The orderly metameric arrangement of dermatomes becomes apparent if we visualize a man in the quadruped position. (From Finneson BE. Diagnosis and Management of Pain Syndromes, ed 2. Philadelphia: Saunders, 1969. Used with permission.)
This is a fairly common occurrence with dental pain. Generally, if the pain is referred to another distribution of the same nerve, it does so in a “laminated” manner. This lamination follows the pattern of the dermatomes. A dermatome is a sensory root field on the skin where pain is felt when a particular neural segment mediates a painful sensation. These have been well charted (Fig 4-3). Each nerve has a corresponding dermatome, but there is considerable overlapping. The upright posture of humans causes some confusion in dermatome arrangement, especially in the extremities. They are better visualized with the subject in the quadruped position (Fig 4-4). This places them in a more logical metameric arrangement and helps considerably to identify the proper segmental relationships. 40
Trigeminal lamination patterns are determined by the manner in which the primary afferent neurons enter in the spinal tract nucleus. According to Kunc,41,42 the location of trigeminal nociceptive terminals within the nucleus caudalis is as follows:
- The fibers from tissues near the sagittal midline of the face terminate highest in the nucleus (cephalic).
- The fibers from tissues located more laterally terminate lowest in the nucleus (caudal).
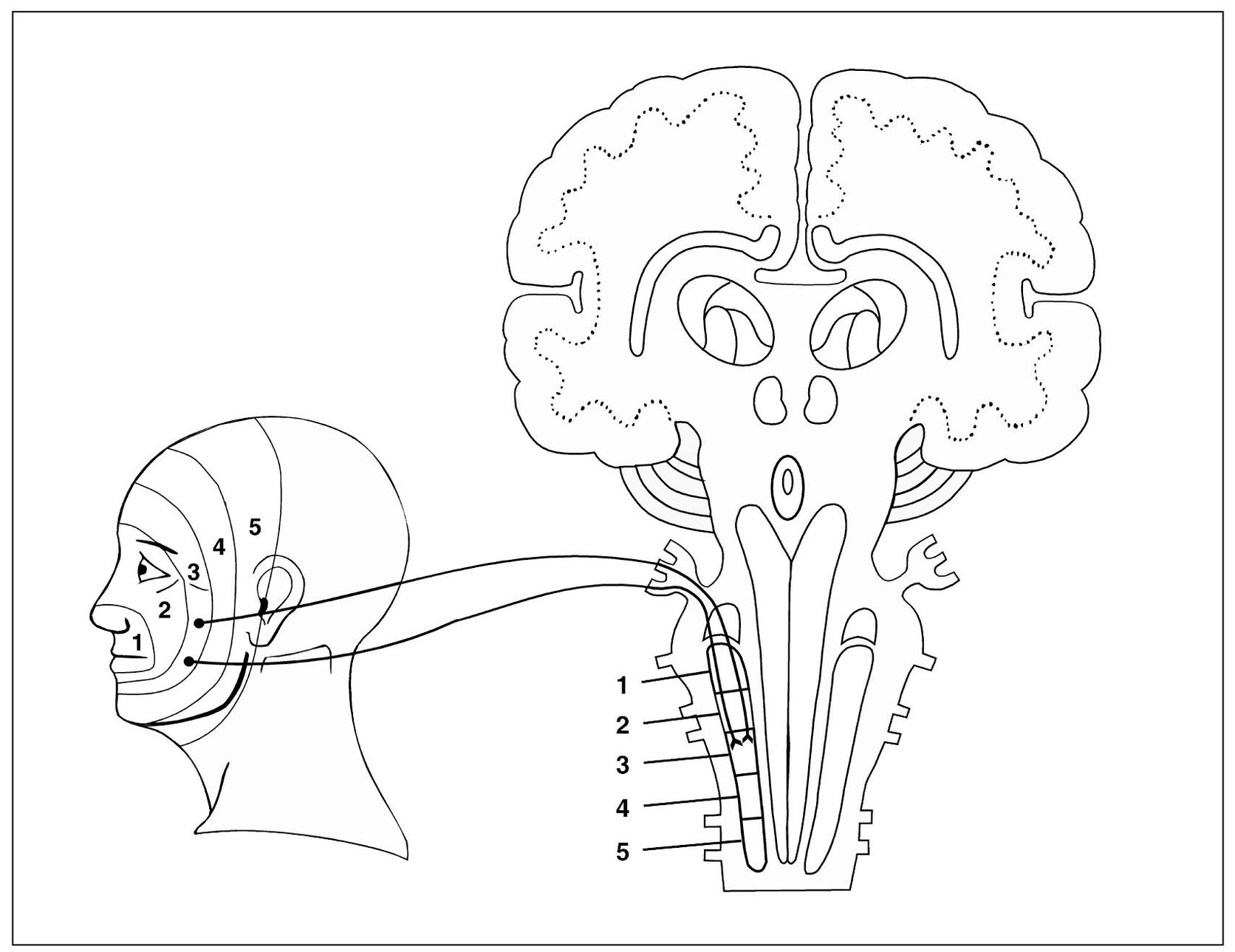
Fig 4-5 Graphic depiction of the spinal trigeminal nucleus and its relationship with incoming sensory input. Note the laminated pattern of innervation from the facial structures into the spinal tract nucleus. These laminated patterns reflect the patterns of referred pain commonly felt in the orofacial structures.
- The intermediate fibers terminate intermediately in the nucleus (Fig 4-5). This laminated arrangement of facial innervation appears to coincide with Finneson’s metameric arrangement of the dermatomes in the quadruped position.40
This grouping of the terminals of the primary trigeminal neurons should influence the location of clinical effects of central excitation, especially with regard to referred pain. It probably accounts for some effects not otherwise readily explained. Kawamura and Nishiyama showed that a molar tooth can project pain to a canine, and a canine can project to an incisor, which confirms the vertical lamination just cited.43
Simply speaking, this means that incisors refer to incisors, premolars to premolars, and molars to molars on the same side of the mouth. In other words, molars do not refer pain to incisors or incisors to molars.
The second clinical rule is that referred pain in the trigeminal area rarely crosses the midline unless it originates at the midline. For example, pain in the right temporomandibular joint will not likely cross over to the left side of the face, nor will right molar referred pain cross to a left molar. However, this is not true in the cervical region or below. Cervicospinal pain can be referred across the midline, although it most commonly stays on the same side as the source.
A third clinical rule of referred pain is that if the referred pain is felt outside the nerve that mediates the pain, it is generally felt cephalic to the nerve (upward, toward the head) and not caudally. Clinically, this means that deep pain felt in the sacral area maybe be referred to the lumbar area, as well as lumbar to thoracic, thoracic to cervical, and cervical to trigeminal. Rarely will trigeminal pain refer to the cervical region. Only very intense primary pain causes excitatory effects in a segment caudal to the site of initiating input.44,45
Secondary hyperalgesia
Hyperalgesia is defined as increased sensitivity to stimulation at the site of pain. Primary hyperalgesia occurs as the result of a lowered pain threshold (sensitization) in the peripheral structures, resulting presumably from the presence of algogenic substances such as bradykinin, potassium, histamine, and 5HT (see chapter 3). Secondary hyperalgesia is different. It is an increased response to stimulation at the site of pain in the absence of any local cause.46 It may occur with or without accompanying referred pain.
Secondary hyperalgesia is also different from referred pain in that it occurs in response to stimulation (provocation) at the site of pain, while referred pain cannot be increased at the site of pain. It is wholly dependent on the source of pain. Both referred pain and secondary hyperalgesia are initiated by deep pain input and remain dependent upon the continuation of such input. They differ, however, in that secondary hyperalgesia persists for a while after the primary pain ceases. Thus, analgesic blocking of the primary pain site does not immediately arrest the hyperalgesia as it does referred pain.
As with referred pain, secondary hyperalgesia may be felt in either superficial or deep structures. Superficial secondary hyperalgesia is felt as sensitive skin, scalp, hair, or gingiva. Deep secondary hyperalgesia is sensed as an area of palpable tenderness, discomfort due to functional manipulation, or hypersensitive or tender teeth.
There are two explanations for the occurrence of secondary hyperalgesia. The first is that related to the sensitization of the second-order neuron. As central sensitization occurs, even normal input can be misinterpreted as noxious.47,48 Therefore, even input carried by the nonnoxious A-beta fibers can be felt as pain. When this occurs, even light touch to the face becomes painful. Pain produced by normally nonpainful stimuli is called allodynia.
A second explanation for secondary hyperalgesia is one that addresses a local cause produced by neurogenic inflammation. As discussed in the last chapter, tissue injury initiates the inflammatory process, which in turn sensitizes neighboring neurons. This is called primary hyperalgesia. With central sensitization, the primary afferent neuron can antidromically release excitatory neurotransmitters (eg, SP) into peripheral tissues, increasing the sensitivity of the primary nociceptors (neurogenic inflammation). When this occurs there can be a small central area of hyperemia, indicating some local tissue reaction. In this flare area of an axon reflex, the pain threshold is lowered, thus indicating that this portion of the hyperalgesic area reflects local tissue change. The entire area of sensitivity, however, extends far beyond the hyperemic zone. The wider area represents true secondary hyperalgesia that is dependent on the continuation of primary deep pain impulses. 45 Analgesic blocking of the source of deep pain that is creating the secondary hyperalgesia does not in fact immediately decrease the total discomfort, presumably because of the flare area that is present.49,50 Anesthetic blocking of the region of primary hyperalgesia will immediately eliminate the pain since the cause is local.
At a clinical level, superficial secondary hyperalgesia usually presents no great problem because there is an obvious lack of local cause. Deep secondary hyperalgesia, however, cannot be distinguished from primary hyperalgesia on the basis of manual palpation or functional manipulation (see chapter 8). Special diagnostic effort is required to make this judgment, which is important therapeutically. It should be noted that palpable tenderness or deep discomfort from functional manipulation identifies only a site of pain, not a source of pain. Whether such an identified site of pain is primary hyperalgesia from a local cause such as inflammation or a heterotopic manifestation of deep pain input located elsewhere is a judgment that must precede definitive therapy.
Efferent (motor) neuron effects
When central excitatory effects alter the function of an efferent neuron, the influence is seen in the muscle innervated by that neuron. There are two effects that may result: muscle co-contraction and the development of myofascial trigger points.
Muscle co-contraction
A common efferent effect secondary to constant deep pain is a reflex excitation of the muscle, which slightly modifies its functional activity51(Fig 4-6). Deep pain input seems to activate a protective response of muscle to limit the movement of the painful part. This response is normal and likely protective in nature. It does not represent pathology. It is interesting to note that, in the presence of deep pain, the antagonist muscles appear to become activated in an attempt to limit activity of the agonist muscle.52,53 This phenomenon is called co-contraction54 and has previously been referred to as protective muscle splinting. The exact mechanisms involved will be discussed in later chapters. It is sufficient to say at this time that deep pains that produce central excitatory effects can influence muscle activity.55
Myofascial trigger points
Another motor effect of central excitation is the development of myofascial trigger points. Myofascial trigger points are characterized by localized areas of hypersensitive bands of muscle tissues found within the body of a muscle. The presence of these trigger points is paramount in the muscle pain condition called myofascial pain.56,57,58 Continued deep pain input of significant intensity can induce the development of myofascial trigger points. The mechanism involved seems to be segmentally related to the site of primary pain. The masseter and temporalis are the masticatory muscles most frequently affected. The trigger point, in turn, refers pain that is frequently felt at or near the site of the initiating primary pain. The trigger itself, however, is usually silent unless manually palpated; then local muscle pain is felt. Although this mechanism may be initiated by pain, once trigger points become established, they remain active or latent until eliminated by therapeutic effort.57,58 Thus, pain at or near the initial site of primary pain may persist as a continuing or recurrent heterotopic manifestation long after the original pain has ceased. The complexity of such pain problems makes accurate diagnosis all the more necessary for effective management of the patient’s complaint. Myofascial trigger point pain will be discussed in more detail in chapter 12.
The muscles that are affected by central sensitization are usually those innervated by the same major nerve that mediates deep pain input, thus establishing a segmental relationship. If the primary pain is mediated by the trigeminal nerve, the muscles most likely to be affected are those innervated by the trigeminal nerve. Eight muscles are innervated by the trigeminal nerve: the masseter, the temporalis, the medial pterygoid, the lateral pterygoid, the mylohyoid, the anterior belly of the digastric, and the tensor muscles of the soft palate and tympanic membrane.
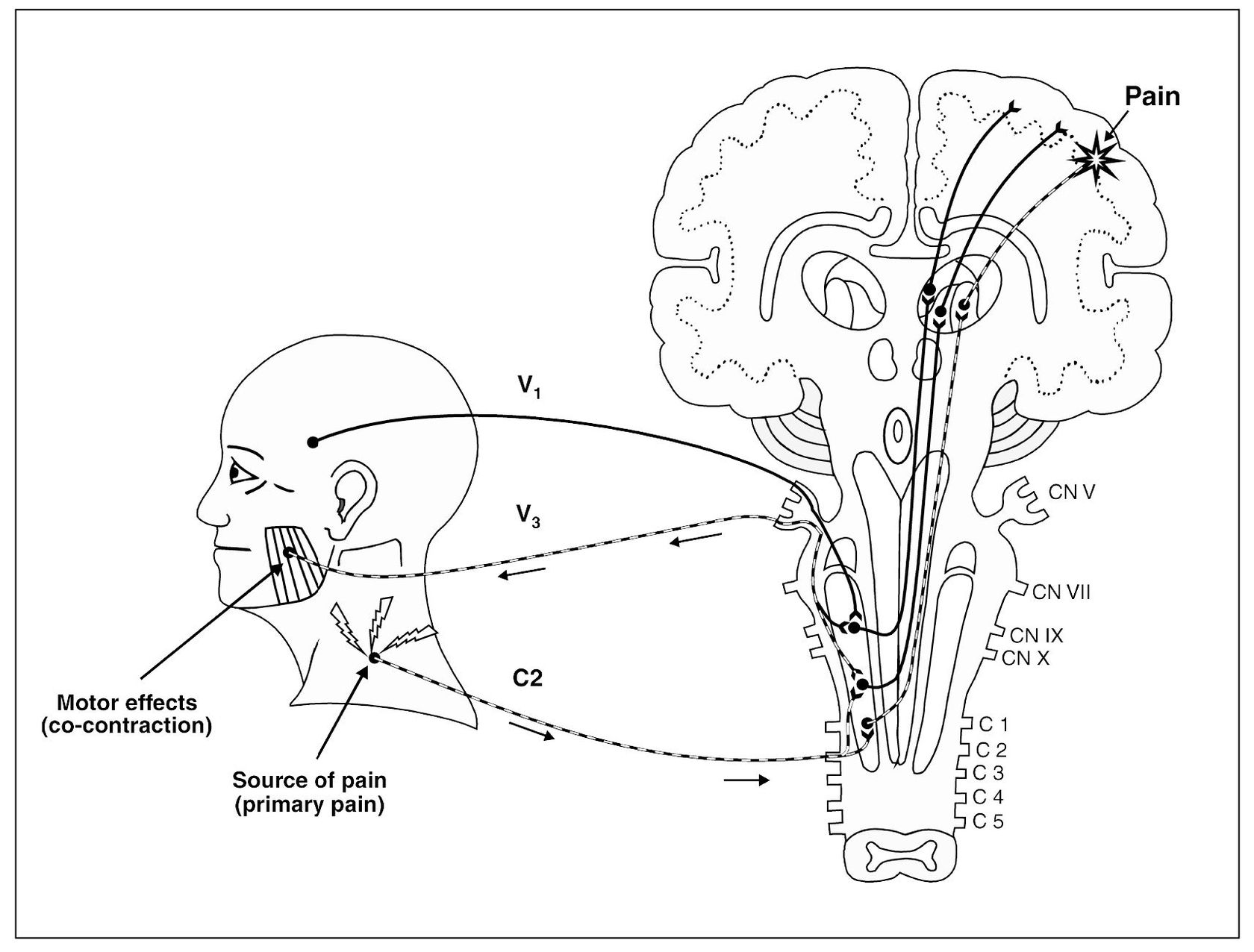
Fig 4-6 Constant afferent nociceptive input can centrally excite efferent (motor) neurons, resulting in co-contraction of associated muscle or the development of myofascial trigger points.
The Experience of Pain
Until recently, human pain was considered a sensory experience evoked by noxious stimulation of neural structures. The impulses thus generated were thought to be transmitted to the CNS, where they were perceived as pain and reacted to. The reaction was thought to comprise extensive behavior on the part of many body systems. The dominant factors in pain reaction were thought to be on a mental level, such as prior conditioning, evaluative significance of the pain, memory, and emotional response. This perception-reaction hypothesis was originated in the 19th century by Marshall59 and Strong.60 It was recognized that great difference in pain reaction was commonplace. Maurice concluded: “The exaggerated reaction is due to psychic factors and is termed psychoneurotic pain.”61 It became popular to think in terms of human discomfort as being organic or psychogenic. Organic pain constituted pain behavior that could be accounted for on the basis of structural conditions. When no structural condition could be found to explain the pain, the clinician labeled it psychogenic pain.
During the second half of the 20th century, there was an increased effort by researchers and clinicians to better understand pain. New experiments and observations gave reason to que/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


