Oral and Dental Care of Local and Systemic Diseases
 Outline
OutlinePrimary Herpetic Gingivostomatitis
Recurrent Herpes Simplex Infection
Fetal Alcohol Spectrum Disorders
Dental and Oral Care Considerations
Patient’s Medical History and Hematologic Status
Hematopoietic Cell Transplantation
Acute and Chronic Oral Complications of Chemotherapy and Radiotherapy
Primary Herpetic Gingivostomatitis
Herpes simplex virus type 1 (HSV-1) is a large DNA virus that causes primary herpetic gingivostomatitis (PHG), mucocutaneous orofacial disease, ocular disease, herpes gladiatorum (seen in athletes involved in contact sports), eczema herpeticum, herpetic sycosis of the beard area, and less frequently herpes genitalis, which is mostly caused by HSV-2.1–3 HSV is one of the most common causes of erythema multiforme.2 Although PHG is a highly contagious infection, only 5% to 10% of patient initially infected develop clinical lesions.4 It is mostly acquired through direct skin contact or bodily fluids, which can spread rapidly in closed settings such as daycare centers and orphanages. The onset is sudden and its clinical manifestations may vary from a mild illness to a severe course requiring hospital admission. Nonspecific symptoms include cervical lymphadenopathy, malaise, irritability, upper respiratory tract infection, and low-grade fever.4 PHG can cause both intraoral and extraoral lesions, and swollen and bleeding gingival tissue. Oral lesions may start as vesicles on the tongue, buccal mucosa, and gingiva, rapidly rupturing to become ulcers 1 to 3 mm in size, which may subsequently form a large ulcerated area covered by a yellowish-grey membrane.4,5 The infection is self-limiting, lasting 10 to 14 days, and healing without scarring.4 Children may present with severe local pain, which can lead to difficulties with fluid and food intake, putting them at risk for dehydration.2,4 Excessive drooling, halitosis, and sore throat are frequently present. Complications are rare but direct transmission may lead to keratoconjunctivitis and herpetic whitlow (e.g., digit, hand, or knee skin infection), which is a classic occupational hazard for health professionals. After the primary infection, the virus may become dormant but can be readily reactivated, producing infections that are generally less severe.1,2,4
Management of PHG is directed at promoting lesion healing, providing palliation, promoting adequate hydration and nutrition, and preventing further spread of the infection through avoiding direct contact with other people and not sharing such items as toys, food, utensils, pacifiers, cups, bottles, toothbrushes, and towels. Drinks and foods with high acid or spice content should be avoided. Cold items such as ice cream, popsicles, and ice chips can soothe affected tissues and help with hydration. Analgesics, topical anesthetics, and coating agents help relieve pain and facilitate food intake, with nutritional supplements added as needed.1,4 Topical anesthetics should be used with extreme caution because they are rapidly absorbed through ulcerated tissues; patients should be encouraged to expectorate after use. Gargling or swallowing anesthetics may lead to suppression of the gag reflex, potentially leading to aspiration. Nonalcoholic antimicrobial rinses may help decrease the risk of secondary infections when there is significant gingival involvement and poor oral hygiene.
Diagnosis is based on the clinical presentation and the patient’s history. However, a positive viral culture is the gold standard. When doing a culture, a swab should be used to soak up the fluid and scrape the base of an unroofed vesicle. The swab is then placed in special viral transport media and sent to the lab. Viral isolates usually grow in tissue culture by 5 days.2 Differential diagnosis include herpangina, aphthous stomatitis, hand-foot-and-mouth disease, Stevens-Johnson syndrome, pemphigus, intraoral zoster infection, necrotizing ulcerative gingivitis, and Behçet syndrome.1–4 There is weak evidence that acyclovir (ACY) can decrease some of the symptoms of PHG and the number of hospitalizations for children under 6 years of age.4 The dosage for oral ACY suspension is 15 mg/kg, with a maximum of 80 mg/kg per day to be used every 3 hours when awake or five times a day for 10 days.2 One of the limitations of the drug is its poor gastrointestinal absorption and bioavailability.3,6 Valacyclovir and famciclovir were developed with an apparently increased bioavailability, but they are not currently available as pediatric suspensions.4
Recurrent Herpes Simplex Infection
Despite the overwhelming success of ACY and related drugs, HSV remains a major global health problem; it is the leading cause of encephalitis and genital ulcerative disease, and a major cofactor for human immunodeficiency virus infection.6 After the primary infection, HSV establishes latency within the trigeminal ganglion. During this period, there is downregulation of the replicative process until a trigger occurs (e.g., illness, sunlight, trauma, emotional stress, menstruation, dental treatment, hyperthermia) leading to viral reactivation, replication in the ganglion, and travel to the skin or mucosal site.3 Asymptomatic shedding, which occurs frequently, is a major risk factor for transmission.3,6 Three forms of HSV infection are recognized: recurrent herpes labialis (RHL), intraoral recurrence, and recurrence mimicking a primary infection.7 Recurrent lesions in immunocompetent patients may occur in the perioral skin, the ala of the nose, and at the junction of the vermilion and the cutaneous lip as RHL.3,7 Intraoral recurrent lesions are seen almost exclusively on the keratinized mucosa of the hard palate and the attached gingiva, and occasionally on the dorsum of the tongue.3,7 In patients with compromised immune systems, recurrent HSV ulcerations may be large, progressive, and persistent (Figure 4-1). The diagnosis is often delayed because of their atypical presentation, which may involve any intraoral site including nonkeratinized areas.3,7
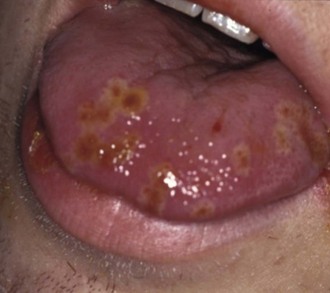
 FIGURE 4-1
FIGURE 4-1Patients who have RHL (cold sores) experience a prodrome of tingling, itching, soreness, and/or burning followed by formation of a papule that progresses to vesicles within hours and to ulceration and crusting in 72 to 96 hours.1,3,5 The highest level of infectivity is the first 24 hours of the appearance of the lesions. Most episodes are self-limited (7 to 14 days) and mild, and because lesions are very short-lived in healthy subjects, the treatment window is also short.1 Recurrences are variable. Lip balms and protective lotions should be used by individuals whose RHL is sun induced.7,8 Symptoms in children may be similar to those of PHG, and palliative management is the same. Virustatic agents have to be started early before a critical mass of virus has replicated, because the resultant damage to epithelial cells cannot be reversed.3 Prophylactic use of antiviral agents for individuals who experience one or two mild episodes a year is not recommended because of the risk of developing resistance and the prohibitive cost relative to benefit.3 ACY has a high selectivity for virally infected cells, presenting minimal toxicity for other tissues. However, its relatively poor oral bioavailability is a major limitation.3,6 Scientific evidence3 suggests that (1) 5% to 10% ACY in ointment base is not efficacious for treating RHL in healthy patients because of poor penetration; (2) 5% ACY in cream base may reduce duration of the lesions if applied early in the prodromal phase, but it does not effectively reduce pain; (3) patients who have more severe and longer episodes are the most likely to benefit from the topical form of the drug; (4) the addition of an antiinflammatory agent may enhance topical ACY action; (5) oral systemic ACY at 200 to 400 mg (for adults) taken 5 times a day for 5 days to treat RHL reduces healing and pain by 1 to 1.5 days as well as shedding, especially if started early; and (6) sunscreen alone (SPF 15 or above) reduces the incidence of RHL.3 Systemic acyclovir (200 mg every 3 hours while awake for 5 days) may be indicated for children with six or more episodes of recurrent HSV per year.8 Other antiviral agents available include valacyclovir, ganciclovir, valganciclovir, penciclovir, famciclovir, foscarnet, and cidofovir. The safety and efficacy of most of these drugs in children have not been established.8 Docosanol, the only over-the-counter drug approved by the Federal Drug Administration (FDA) for the treatment of RHL, can be used in children older than 12 years. It is applied five times daily to the affected area of the face and lips. The medication should be started at the earliest sign or symptom to obtain maximal efficacy.3,8 Many other antiviral agents and vaccines are being tested and developed, including imiquimod, resiquimod, undecylenic acid, thymopentin, and mucosally delivered microbicides.3,6
Patients undergoing cancer treatment have a high risk for development of recrudescent HSV infections, thus their management is toward prophylaxis.3 In this group, lesions that do not heal within 7 to 10 days should be recultured or rebiopsied, and sensitivity testing to ACY be performed.3 Individuals who have received a hematopoietic stem cell transplant have shown the highest rate of ACY resistance, which is treated with foscarnet.3 Patients who have compromised renal function should have ACY doses reduced to about 50%.3 In cases of both PHG and recurrent HSV infections, the dental professional should address proper nutrition, hydration, optimal oral hygiene, local control of the process, and cleanliness to prevent autoinoculation and spread to others. Dental treatment should be avoided until healing occurs.
Oropharyngeal Candidiasis
Candidiasis is an opportunistic infection caused by the overgrowth of Candida species due to antibiotic and/or corticosteroid use, xerostomia, diabetes mellitus, appliances covering the palate, smoking, and/or an immature or compromised immune system.8,9 The most common forms in children are pseudomembranous (Figure 4-2) and erythematous candidiasis. The former appears as white to yellow movable papules and patches of various sizes and shapes, growing in a radial pattern and concealing the underlying erythema.8,10,11 It typically occurs on the buccal mucosa, mucobuccal folds, dorsolateral tongue, and oropharynx.10 The erythematous or atrophic form ranges from a diffuse to patchy redness involving mostly the palate and dorsum of the tongue.8,10 Accumulation of saliva in deep folds of the corners of the mouth allows for colonization of fungal and bacterial organisms (angular cheilitis)10 (Figure 4-3). It is red, sometimes ulcerated and crusted, usually painful, and more common in children with a lip sucking habit, which may lead to spread of the infection to the adjacent perioral skin.10 Patients with oral candidiasis can experience a painful mouth, a burning feeling on the mucous membranes and tongue, dysphagia, and difficulty eating or drinking.11 Diagnosis is reached through a consideration of patient demographic and health factors, clinical signs and symptoms, and laboratory criteria. A thorough assessment of the patient’s health and current medications is critical to ensure effective antifungal therapy and to decrease the frequency of recurrences. Cultures are very helpful in cases where the lesions are resistant to therapy.
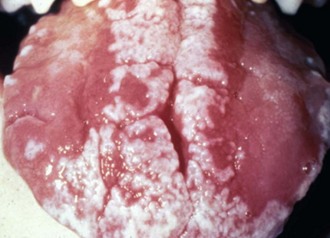
 FIGURE 4-2 Pseudomembranous candidiasis.
FIGURE 4-2 Pseudomembranous candidiasis.
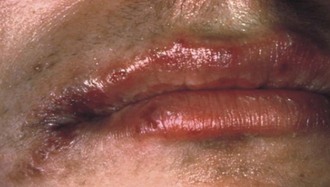
 FIGURE 4-3 Angular cheilitis.
FIGURE 4-3 Angular cheilitis.
It is important to improve oral hygiene, control caries, keep pacifiers and appliances clean, and replace contaminated toothbrushes to control disease.8 Most available antifungal agents have reasonably broad activity against Candida albicans and other Candida species, which are more commonly isolated. Unexplained or frequent relapses should prompt an evaluation for occult systemic disease, conditions that can lead to an immunocompromised state, or maternal breast in breastfeeding infants.11 Acquired resistance has been described in Candida isolates and is most often associated with long-term use of antifungal agents.12 Topical antifungal agents include compounded clotrimazole suspension (10 mg/ml) and nystatin oral suspension (100,000 U/ml) to swish for 2 minutes and swallow or expectorate four times daily for 2 weeks, followed by a reevaluation of the oral cavity.8,11 The patient should not eat or drink for 30 minutes afterward. Adolescents can use one to two pastilles (200,000 U) slowly dissolved in the mouth five times daily.10 Nystatin solution contains 30% to 50% of sucrose so oral hygiene must be reinforced. Clotrimazole troches (10 mg), also very rich in sucrose, can be used by slowly dissolving one troche in the mouth every 3 hours while awake (five per day) for 14 days.8 Systemic antifungal drugs are advantageous when other topically delivered medications are administered concurrently. They include fluconazole 6 mg/kg orally every 12 or 24 hours for 5 to 7 days; adolescents can use a 200-mg loading dose and then 100 to 200 mg once a day for about a week.10,11 Ketoconazole may also be used in children at 5 to 10 mg/kg every 12 or 24 hours, and in adolescents 200 to 400 mg every 24 hours for 5 to 7 days.10,11 It is highly effective and has the advantage of good patient compliance. Common side effects with systemic use include nausea, vomiting, pruritus, skin rash, abdominal discomfort, headache, abnormal liver function tests, and drug-induced hepatitis.10 Chlorhexidine gluconate 0.12% can be used as an antimicrobial rinse and is most useful for maintenance purposes rather than as a first-line antifungal agent.8 Antifungal ointments and creams include nystatin, clotrimazole, miconazole, and ketoconazole. For chronic cases of angular cheilitis, nystatin and triamcinolone acetonide cream (Mycolog-II) is the best choice when applied to the corners of the mouth three times a day for 5 days.8
Invasive fungal infections in children are on the rise because there has been an increase in the number of children with primary and secondary immune deficiencies.12 These infections tend to occur in hospital settings and are mostly caused by Candida spp., followed by Aspergillus spp., which cause the most common mold infection.12–14 Presence of Candida in the blood (candidemia) in children is associated with high morbidity and mortality, increased length of hospital stays, and higher health care costs.14 The infection can disseminate to end organs, including brain, lungs, liver, kidneys, eye, and spleen. Risk factors for candidemia include prolonged stay in an intensive care unit, immunosuppression, prior bacterial infection, recent surgery, presence of a central venous catheter, mechanical ventilation, dialysis, extended vancomycin use, diabetes, trauma, and total parental nutrition.13,14 It can be treated with fluconazole or an echinocandin for empirical therapy in suitable candidates.12,14 Caspofungin is the only echinocandin currently approved for use in children by the FDA, but its half-life in children is one third less than in adults.14,15 Children, and especially neonates, tolerate conventional amphotericin B better than adults. However, pediatric patients need higher doses of voriconazole, the drug of choice for invasive aspergillosis and many other mold infections in both children and adults, to achieve therapeutic levels.12,15 It is also an effective therapy for candidiasis.15 There is little current data on the pediatric use of second generation triazoles (such as posaconazole, isavuconazole, ravuconazole, and albaconazole), and other echinocandins (anidulafungin, micafungin).12 Itraconazole can be used in children, but it is not nearly as easy to administer and monitor as fluconazole.15 Despite the advent of new antifungal drugs, invasive fungal infections, including those caused by Candida spp., remain a serious problem due to limitations of toxicity, high treatment cost, and resistance.13
Interestingly, the benefits of prophylaxis to reduce invasive disease for children with hematologic malignancies, bone marrow or solid organ transplantation, or primary immune deficiencies are not clear. Because there have been no significant randomized, controlled trials evaluating fungal prophylaxis in any of these settings, the routine use of prophylactic agents is not recommended, except for itraconazole prophylaxis in children with chronic granulomatous disease.12
Sickle Cell Disease
Sickle cell anemia (SCA) is the most common genetic disorder of the blood and is most frequently observed in persons of African, Afro-Caribbean, Middle Eastern, Indian, Central and South American, and Mediterranean ancestry.16,17 SCA comprises sickle cell trait, which is benign, and sickle cell disease (SCD), which is a group of disorders characterized by hemolysis, chronic organ damage, and unpredictable acute complications that may become life-threatening.16 It is caused by a variant of the B-globin gene called sickle hemoglobin (HbS), which is involved in oxygen transport. The molecular nature of the disease is a substitution of valine for glutamic acid at the sixth amino acid in the B-globin protein, which allows HbS to polymerize when deoxygenated.18–20 In the United States, it is the most prevalent disorder identified by neonatal screening.16,17,20,21
The polymerization of deoxygenated HbS is an indispensable event in the pathogenesis of SCD and is dependent on the HbS concentration in red blood cells (RBCs), the degree of cell deoxygenation, and the pH and intracellular concentration of fetal hemoglobin (HbF), which is protective against sickling.19 The process occurs after a period of time necessary for sufficient intracellular polymerization to deform the cell. The resulting polymer is a ropelike fiber that aligns with others, forming a bundle and distorting the RBC into a sickle shape, which interferes with their deformability. The sickled cells are trapped mostly in the slow-flowing venular side of the microcirculation, enhancing their adhesion to the endothelium, forming a heterocellular aggregate that leads to local hypoxia, increased HbS polymerization, and spread of the occlusion to the adjacent vasculature.19 Thus, HbS produces a problem of RBC “sticking” rather than simply sickling, leading to chronic endothelial damage. The remarkably varied clinical picture is one of a chronic inflammatory vascular disease with evolving organ damage punctuated by periods of severe pain and pulmonary complications.18,19,22,23 Anemia and vasculopathy are the hallmarks of the disease, with symptoms appearing within the first 6 months of life.17,20 The mean life span of a sickled RBC is reduced from 120 days to 12 to 17 days, Hb levels are 6 to 9 g/dl (normal: 12 to 18 g/dl), and reticulocyte counts are 5% to 15% (normal: 0.5% to 1.5%).18
Painful crises in early years manifest as dactylitis (hand-foot syndrome) and can be triggered by infection, dehydration, extreme temperatures, hypoxia, physical or emotional stress, and menstruation.18 Bone pain is often excruciating, symmetric, and present in multiple locations, lasting from a few minutes to several days.19,23 Splenic sequestration crises are caused by a large number of RBCs becoming trapped in the spleen and can induce sudden and severe anemia, thrombocytopenia, and reticulocytosis.19,20 Life-threatening postsplenectomy sepsis is primarily caused by polysaccharide encapsulated bacteria, particularly Streptococcus pneumoniae, which is a leading cause of mortality among infants affected by SCD.23–25 Other systemic manifestations include cardiovascular problems, osteomyelitis, osteoporosis, growth disturbances, osteonecrosis, acute chest syndrome, cerebrovascular accidents, chronic renal failure, and priapism. Psychosocial complications, including impaired psychosocial functioning and decreased quality of life, are not uncommon.26
The most important intervention in SCD is administration of penicillin V potassium 125 mg orally twice daily starting at 2 months of age to prevent pneumococcal infection. The dose is doubled at age 3 years and should be continued until age 5 years. It should also be given for 2 years following splenectomy.16,24,27 Much research is being done to find the ideal drug to treat the disease. Hydroxyurea (HU), initially used as an antineoplastic agent, is associated with increased HbF levels, which leads to fewer sickling episodes and fewer long-term sequelae.28 Its mild and transient toxicities include anemia, nausea, neutropenia, and thrombocytopenia. The only available cure for the disease is hematopoietic stem cell transplantation, which should be done at an early age before organ dysfunction develops.19,21,28,29 In general, mortality has decreased, the mean age at death in the United States has increased (42 years for men and 48 years for women homozygous for HbS), and fewer patients die of infection.22,25
Oral and Dental Manifestations
The oral and dental manifestations of SCD are nonspecific and may be present in healthy individuals as well as in those with other systemic disorders. The mucous membranes may show jaundice due to hemolysis and pallor caused by a low hematocrit. Glossitis and delayed tooth eruption may also be apparent.30,31 Radiographic findings include decreased radiodensity in the bones, coarse trabecular pattern, thin inferior border of the mandible, loss of alveolar bone height, pronounced lamina dura, dentin hypomineralization, interglobular dentin in the periapical region, calcifications in the pulp chamber, and hypercementosis.31 Craniofacial abnormalities include bimaxillary protrusion with flared incisors, prominent parietal and zygomatic bones (“tower skull”), widening of the diploic space with thinning of the outer table of the calvarium and vertical trabeculations (“hair-on-end” appearance), and fibrotic calvarial lesions with a ringlike appearance (“doughnut lesions”).31,32
Sickling crises within the microcirculation of the facial bones and dental pulps may cause orofacial pain without any odontogenic pathology.33 Vasoocclusive episodes near the mental nerve foramen may result in persistent paresthesia of the lower lip.33 SCD patients are not more prone to periodontal disease than the general population. It seems that long-term use of penicillin prophylaxis in these patients prevents the acquisition of mutans streptococci, resulting in significantly lower caries rate in the primary dentition. However, by 8 years of age, once the prophylactic regimen has stopped, children experience the same level of caries as unaffected peers.34
Oral and Dental Treatment Concerns
Individuals carrying the trait present no challenges for dental treatment. For patients affected with the disease, a detailed medical history must be taken, including related complications, characteristics of the pain crises, past and current medical care, presence of orthopedic prostheses, carriage of blood-borne viruses, growth and development issues, medications (including use of bisphosphonates), allergies, and a brief psychological profile.35 The patient’s dental history should be reviewed and the family educated about the role of optimal oral health to increase quality of life. Complaints of pain in healthy teeth must be taken seriously because the possibility of pulpal infarction and necrosis exists with SCD. No conclusions have been drawn regarding the best approach in these situations. All oral infections must be treated vigorously, and elective surgeries should be avoided.30 Orthodontic planning should include consideration the bony architecture and physiology.30,31 If the patient is using or has used bisphosphonates, the family must be informed of the risk of bisphosphonate-related osteonecrosis of the jaws following invasive dental procedures. All dental care can be safely done in the dental office during noncrisis periods. Individuals who have orthopedic prostheses may require antibiotic prophylaxis before invasive dental procedures.21,24 If the patient is taking HU, a complete blood count is warranted because of the risk of neutropenia and thrombocytopenia.35 Coagulation factors should also be checked if the liver is involved. There is no evidence to support the use of local anesthetics without vasoconstrictors in SCD patients.31 Use of nitrous oxide is safe but care should be taken to avoid hypoxia. Oral sedation can be considered after a consultation with the patient’s hematologist and careful selection of drugs that do not lead to respiratory depression. An experienced anesthesiologist should be consulted before proceeding with dental care under general anesthesia in order to determine the patient’s risk of complications and to prevent perioperative problems.35 Low-risk patients can be treated in an outpatient surgery center; higher risk individuals need a fully equipped operating room in a hospital facility for adequate medical support. The National Institutes of Health do not recommend simple transfusions for dental or oral surgery procedures.18,21
Hemophilia A
Hemophilia A is an X-linked recessive disorder resulting in deficiency of plasma factor VIII coagulant activity, occurring in 1 in 5000 male births. It accounts for about 85% of all hemophilias and can be classified as severe (plasma has no detectable factor VIII), moderate (only 1% to 4% of the normal factor VIII level is present), and mild (5% to 25% of the factor is present). In mild cases there may be no history of bleeding, which may only be observed after trauma or surgery. The condition should be suspected when a male patient presents unusual bleeding with a normal platelet count, bleeding time, thrombin time, and prothrombin time, but a prolonged activated partial thromboplastin time.36–42 Specific factor assays are needed to differentiate between hemophilia A and B (factor IX deficiency), which accounts for 10% to 15% of all hemophilias.
The clinical hallmarks of hemophilia A are joint and muscle hemorrhages, easy bruising, and prolonged and potentially fatal hemorrhage after trauma or surgery, but no excessive bleeding after minor cuts or abrasions.36,40,42 Hemarthroses are first seen when the child begins to walk. If the joint hemorrhages go untreated, severe limitation of motion occurs and may lead to permanent disability. Intramuscular hematomas can compress vital structures and may cause nerve paralysis, as well as vascular or airway obstruction. Clinical management of hemophilia A is done according to the severity of the condition, type of bleeding present or anticipated, and presence of inhibitors.36,40,42–44 Regular infusions of factor VIII should be used in cases of major surgery and life-threatening bleeding. Vasopressin (desmopressin) increases the plasma levels of factor VIII an/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


