CHAPTER 3 Pharmacotherapeutics
The Clinical Use of Drugs
As stated in Chapter 1, drugs are often selective in the effects they produce because they activate or inhibit specific drug receptors. Even the most selective agents generally evoke a spectrum of reactions, however, rather than a single pharmacologic outcome. Atropine in therapeutic concentrations specifically prevents the stimulation of muscarinic receptors by acetylcholine. Because these receptors are vital to the normal function of the entire parasympathetic nervous system, their blockade can result in a wide range of autonomic responses. Although specific in action, atropine is nonselective in effect. In addition, specificity of receptor binding is usually a matter of dose; in concentrations greater than therapeutic, atropine blocks the nonmuscarinic effects of acetylcholine and may inhibit the actions of other chemicals, such as histamine and 5-hydroxytryptamine. Finally, nonspecific effects unrelated to receptor blockade may be observed. Large concentrations of atropine have local anesthetic activity and directly affect the central nervous system (CNS) and peripheral vasculature.
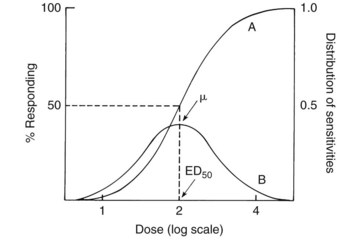
In addition to the fact that single agents can produce multiple effects, pharmacotherapeutics is complicated by variations in patient responsiveness. A therapeutic dose of drug for one person may be ineffective for a second person and toxic to a third person. Even highly inbred laboratory species display measurable biologic variations in drug sensitivity. Figure 3-1 is a quantal dose-effect graph illustrating the percentage of subjects responding to an agent as a logarithmic function of the dose. The graph is constructed by counting the number of animals or patients exhibiting a specified effect at various doses. With low amounts of drug, very few individuals react; as the dose is increased, however, more are affected until a dose is reached at which the response is universal. Although similar in appearance, this quantal dose-effect relationship must not be confused with the graded dose-response curve described in Chapter 1 (see Figure 1-6). The quantal dose-response curve is sigmoidal because of the log-normal distribution of drug sensitivities found in most populations (see Figure 3-1). The median effective dose (ED50) is the amount of drug required to produce a particular effect in 50% of treated individuals. Although potency is represented in quantal and graded relationships by the position of the curve on the abscissa, intrinsic activity or efficacy is apparent only in graded responses. Biologic variation, which is inversely correlated with the slope of the quantal dose-effect curve, cannot be estimated from a single graded dose-response graph.53
FACTORS INFLUENCING DRUG EFFECTS
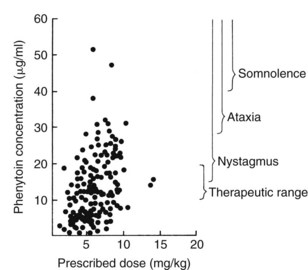
Differences between patients in reaction to a therapeutic agent may arise from disparities in drug concentration obtained with a standardized dose (pharmacokinetic differences), from variations in individual responsiveness to a given drug concentration (pharmacodynamic differences), or from secondary factors such as the failure of patients to take their medication as prescribed (noncompliance). Figure 3-2 shows the lack of correlation that can develop clinically between the prescribed dose of a drug—in this case the anticonvulsant phenytoin—and the resultant plasma concentration and pharmacologic response. Even with the daily dose corrected for body weight, this study revealed that the steady-state concentration of phenytoin differed 20-fold or more.27 A small percentage of patients experienced nystagmus, an early indication of drug toxicity, at plasma concentrations barely sufficient to control convulsions in other patients. It is apparent that, given a therapeutic concentration range of 10 µg/mL to 20 µg/mL (the plasma concentration of phenytoin supposed to provide seizure protection with minimal adverse effects), most patients were prescribed or took on their own either insufficient medication or an overdose. Although pharmacokinetic dissimilarities account for many differences in patient responsiveness, the fact that phenytoin has a “therapeutic range,” rather than a single effective concentration, indicates that there also exists some variation in pharmacodynamic sensitivity to the anticonvulsant.
Patient Factors
Age
Pediatric patients generally cannot be given adult dosages of drugs. The primary reason is their smaller body size, and various formulas (discussed in Chapter 55) have been devised to calculate pediatric fractions of the adult dose. For the following reasons, however, children must not be thought of as merely miniature adults. First, even with the size differential taken into account, neonates display an unusual hyperreactivity to drugs. Immature hepatic and renal systems during the first weeks of life tend to promote drug accumulation, and the relative inefficiency of drug binding by albumin (sometimes because of competition for binding sites by bilirubin) may also lead to abnormal concentrations of drug in the vicinity of receptors. In addition, distribution of compounds into the CNS may be enhanced by an incomplete maturation of the blood-brain barrier. Second, in contrast to neonates, children and infants older than 6 months often require large milligram-per-kilogram body weight doses of drugs during therapy. This relative hyporeactivity is mostly attributable to an enhancement in the rate of elimination.14 Dosage adjustment on the basis of surface area (see Figure 55-6) rather than body weight is empirically a useful strategy in correcting for age-related differences in elimination.
Geriatric patients are frequently hyperreactive to drugs. Although increased sensitivity may result from organic pathologic conditions or from drug interactions (both more likely to occur in elderly patients), age-related functional changes in drug disposition and cellular responsiveness are also involved. Because patients older than 65 years are much more likely to experience adverse drug reactions than young adults, at least in part because elderly patients consume many more medicines, careful selection of drug and dosage schedules is necessary, especially with drugs of low safety. Geriatric pharmacology is becoming increasingly important to the dentist as the general population ages and a higher proportion of elderly individuals retain their teeth (thanks to improved oral hygiene and professional care); this subject is covered in its entirety in Chapter 53.
Sex, pregnancy, and lactation
Women seem to be more susceptible to drug-induced blood dyscrasias, and women taking systemic contraceptives may be more prone to some drug interactions. Drug-induced torsades de pointes is a potentially life-threatening arrhythmia with a significant sex bias. Women may be more likely to develop torsades because the QT interval of the electrocardiogram (see Chapter 24) is longer in women after puberty. The antiarrhythmic sotalol, one of the approximately 50 drugs that prolong the QT interval, is associated with a three times higher incidence of torsades in women.7 Because the preapproval clinical trials of sotalol enrolled only men, the relatively common side effect of QT prolongation was not recognized before the drug was released for general use.
Pregnancy is a major concern in pharmacotherapeutics. Alterations in liver function are common, and the hepatic toxicity of tetracycline and certain other compounds is markedly accentuated by pregnancy. The metabolism of numerous drugs is increased because of the ability of the high estrogen and progesterone concentrations to stimulate the pregnane X receptor (see Chapter 2) and cause enzyme induction. Renal excretion is likewise increased because of the elevated cardiac output and glomerular filtration. When present, pregnancy toxemia may increase drug effects by reducing the binding capacity of albumin, which is already reduced in a healthy pregnancy.
Environmental factors
Numerous chemicals that are ingested, inhaled, or absorbed through the skin can influence the body’s disposition of, or response to, various drugs. Patients receiving monoamine oxidase inhibitors risk severe hypertension and death if they eat foods containing tyramine (e.g., certain cheeses, beers, and wines). The therapeutic effects of levodopa in parkinsonism may be prevented by pyridoxine (vitamin B6), present in foods and multivitamin supplements. Grapefruit juice contains substances that inhibit the CYP3A enzymes responsible for metabolizing a host of drugs (see Chapter 2). Finally, the use of insulin must be carefully matched to the patient’s dietary intake to avoid complications associated with hypoglycemia and hyperglycemia.
Physiologic variables
Many physiologic functions reveal a daily periodicity of intensity. These circadian rhythms often result in daily fluctuations of drug responsiveness. In dentistry, the duration of local anesthesia after nerve blockade varies by a factor of two during the course of a day, with the greatest effect occurring in the afternoon in patients with normal sleep patterns.40
Pathologic factors
Hepatic dysfunction, whether caused by specific hepatic disease, infection, or other conditions, can markedly retard the metabolism and biliary excretion of drugs. Reduced transport capabilities can inhibit the uptake of drugs into the liver and export of metabolites from it.52 Standard liver function tests are of little prognostic value regarding drug biotransformation. Some patients with demonstrable cirrhosis or hepatitis may show little metabolic deficit, whereas others may exhibit marked hyperreactivity to standard doses of drugs. Within the same individual, the metabolism of some drugs may be impaired but not others. Because the liver is responsible for the synthesis of plasma proteins such as albumin and pseudocholinesterase, and for the breakdown of compounds such as bilirubin that compete for drug binding sites in plasma and various tissues, hepatitis may significantly alter (up or down) a drug’s volume of distribution and elimination half-life independently of its specific effects on hepatic drug metabolism. For drugs with high hepatic clearance, metabolism is decreased by cirrhosis-induced reductions in total liver blood flow. The uncertainties of drug metabolism introduced by hepatic disease require that substances inactivated in the liver be used cautiously in affected patients and that drug effects be monitored carefully to avoid serious adverse reactions.
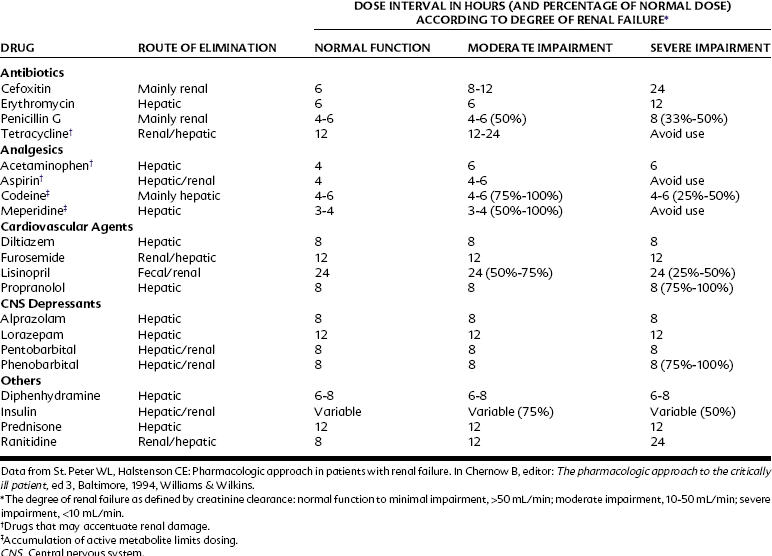
Renal disease is a common modifier of drug effects. The plasma half-lives of agents eliminated in the urine are often greatly prolonged by renal failure. Even for compounds completely inactivated in the liver, inadequate excretion of metabolites may increase the incidence of untoward reactions. A good measure of renal status is provided by the endogenous creatinine clearance. A 50% decrease in creatinine clearance should theoretically indicate a twofold increase in the elimination half-life of a drug that is removed from the blood solely by glomerular filtration. For a drug partially eliminated in the urine, the increase in plasma half-life should be correspondingly less. The customary approach to avoiding excessive drug accumulation in patients with renal disease is to lengthen the dosage interval in accordance with the degree of impaired elimination. Table 3-1 lists for several drugs (including some commonly used in dentistry) the approximate dosage intervals indicated for patients with moderate or severe renal failure.44 Although it is possible for active secretory and resorptive processes to be relatively less or more affected by renal disease than is glomerular filtration, the renal elimination of most drugs simply parallels the creatinine clearance.
An insidious form of interaction between pathologic factors and drug effects occurs with agents potentially toxic to their primary organs of elimination. Acetaminophen accumulation permitted by liver disease may result in hepatic necrosis and further impairment of drug metabolism.10 A similar vicious cycle involving the kidney has been observed with various drugs.
Genetic influences
Genetic variables contribute greatly to the differences in drug responsiveness illustrated in Figure 3-1. Although the importance of heredity is underscored by the evolution of pharmacogenetics into a recognized field of study, the elucidation of multigenetic factors that lead to log-normal distributions in drug reactivity has proved difficult (see Chapter 4). Previously, the only variations in drug effects that had been unequivocally linked to genetic differences were variations that exhibit simple inheritance patterns and yield bimodal or otherwise discontinuous distribution curves and variations that can be associated with certain groups of people on the basis of blood type, race, or ethnic background. Now, studies of gene expression and polymorphisms are helping to uncover an increasingly broad array of genetically determined differences in drug responsiveness. Genetic factors are responsible for idiosyncratic reactions and determine, in part, the relative likelihood of a patient having other adverse responses to an administered agent. Genetic influences can alter drug effects quantitatively; they may also result in the appearance of novel pharmacologic outcomes. Genetic influences affecting drug metabolism and drug receptors are discussed in Chapter 4.
Drug Factors
Variables in drug administration
Of all factors influencing pharmacologic responses clinically, only those involved with drug selection and administration are totally under the control of the clinician. Some of these variables—dose, drug formulation, route of administration, and drug accumulation—are discussed in detail in previous chapters. Two additional factors are the time of administration and the duration of therapy. Many disturbing side effects are minimized if an agent can be given shortly before sleep, including the autonomic effects of the belladonna alkaloids, the vestibular component of nausea associated with opioid analgesics, and the sedative properties of the antihistamines. Conversely, agents producing mild CNS stimulation are better tolerated in the daytime. The scheduling of doses with or between meals to limit gastrointestinal upset or to enhance absorption is discussed in Chapter 2.
Drug tolerance and sensitization
Although the bases for most types of cellular tolerance are not well understood, it seems that adaptive changes to oppose drugs administered long-term that act on specific receptor systems are a common phenomenon. Receptor responsiveness is not static; in the case of agonist drugs, receptors may become diminished in activity and number through the respective processes of desensitization and downregulation, as described in Chapter 1. Response elements downstream from the primary receptor may be similarly affected. Other adaptive changes may include alterations in endogenous mediator synthesis, storage, release, and reuptake.
A final form of tolerance involves drug-induced changes in cellular distribution. A classic example is the development of tolerance to anticancer chemotherapy by cells that overexpress the drug exporter P-glycoprotein, otherwise known as multidrug resistant protein-1. Similarly, the induction of P-glycoprotein by toxins may increase the ability of the blood-brain barrier to prevent their entry into brain cells.30 This form of “distributional tolerance” exhibits aspects of pharmacokinetic and pharmacodynamic tolerance—pharmacokinetic in the sense that the distribution of the drug is altered and pharmacodynamic in that increasing the dose may be an unsuitable strategy to restoring drug effect (because of toxicity to cells not protected by P-glycoprotein).
Pharmacodynamic sensitization, in which the individual becomes increasingly responsive to drugs administered on a regular basis, has been documented for several CNS stimulants. Cocaine given in single daily doses to rats causes increased motor activity after 1 week of treatment. This effect, associated with increased release of dopamine in the brain, is a conditioned response because placebo substitution for cocaine after 1 week elicits a similar response.19
Factors Associated with the Therapeutic Regimen
Placebo effects
A placebo effect is any effect attributable to a medication or procedure that is not related to its pharmacodynamic or specific properties.55 The term placebo is derived from the Latin verb placere, meaning “to please.” In pharmacotherapeutics, a placebo may be either “pure,” in which the preparation is pharmacologically inert (e.g., a lactose tablet), or “impure,” in which the drug has pharmacologic activity, but is given for a condition or in a manner such that no benefit can be obtained from its specific properties. Two commonly held misconceptions are that placebos provide nothing more than a means of placating patients and that they may help in psychosomatic illness but are worthless when symptoms are organically based. Numerous studies have revealed, however, that placebo medication is effective in treating the subjective responses to various “real” medical conditions (e.g., the pain of cancer, angina pectoris, headache, and surgical wounds). The distinction between psychogenic and organic illness has become blurred by the realization that psychological disturbances often produce physiologic or pathologic manifestations and that organic diseases, or at least their signs and symptoms, can be influenced by the CNS through regulation of hormonal secretion and peripheral nervous system activity. Placebo effects are not merely subjective in nature; the administration of pharmacologically inert substances has led to measurable changes in gastric acid secretion, in heart rate and blood pressure, in the number of circulating leukocytes, and in the plasma concentrations of various compounds, including adrenal steroids, catecholamines, electrolytes, and glucose. Even so-called subjective responses to placebos may have a biochemical basis. It has been argued that placebo analgesia can be blocked by naloxone, a specific opioid antagonist,24 and that the placebo effect may involve the dopaminergic reward system.12 Nevertheless, “physical parameters” (e.g., blood pressure and bronchial muscle tone) are much more likely to be affected than are “biochemical parameters” (e.g., blood cholesterol or glucose concentrations).42
Placebo responses to drugs arise from expectations by the patient concerning their effects and from a wish to obtain benefit or relief. Expectations develop at the conscious and subconscious levels and are influenced by many factors. The patient must be aware that treatment is being rendered. The symbolic association of receiving medication in a therapeutic environment generates placebo reactions. The patient must also be anxious about the problem and desirous of being cured. Placebo effects are unlikely if there is patient indifference to the condition or to the therapeutic regimen. Past experience is another important variable. Previous drug exposure informs a patient of what to expect from a drug; repeated administrations evoking prompt, noticeable effects may produce conditioned reflexes. Because suggestion is involved, placebo effects are subject to modification by the practitioner’s attitudes (toward the patient, toward the patient’s illness, and toward the drug or placebo) and how these feelings are communicated. In one study, a 45% reduction in placebo response occurred solely as a result of the administrators’ negative bias toward the placebo medication.3
More recently, the placebo effect has been more carefully scrutinized. Because most studies using placebo controls have not adequately distinguished between the effects they produce and the natural course of a symptom, disease, or healing process, the placebo effect may have been overemphasized.18 The administration of a placebo involves the placebo intervention (e.g., the giving of a lactose tablet) and the remainder of the patient-doctor interaction.17 Determining the relative contributions of each experience may be especially difficult.
Placebos are valid and often necessary inclusions in clinical trials, especially in studies such as analgesic drug trials, in which the placebo effect is well documented.15 Studies involving other subjective outcomes also represent a strong argument for placebo use.30 Placebo effects advantageous to therapy—in addition to beneficial pharmacologic effects—should also be sought whenever a drug is administered clinically.33 Sometimes the effective communication of confidence and other positive attitudes by the practitioner can make the difference between therapeutic success and failure. The clinical application of placebo drugs should be restricted, however, to conditions for which no other agent is superior. Even then, the evolution of informed consent into a basic patient right has, at best, complicated the clinical administration of placebo medication.33 Despite the apparently widespread continuing use of placebos (mostly impure varieties) in medicine,49 there seems to be no justification for the therapeutic use of placebo medication in routine dental practice.
Medication errors and patient noncompliance
Medication errors commonly result in suboptimal therapy and occasionally life-threatening responses. Poor pharmacotherapeutic decisions by the clinician may stem from a lack of knowledge about the patient, disease, or drug. In addition, drugs are often not used in the manner intended by the prescriber. Occasionally, the clinician may miswrite the prescription, or the pharmacist may supply the wrong drug or incorrectly transcribe the instructions to the patient. In the hospital setting, the nursing or house staff may administer the drug incorrectly, neglect to administer it, or administer it to the wrong patient. Most medication errors arise, however, from the failure of patients to take their preparations as directed. Drug defaulting is a major problem in therapeutics; most studies document a noncompliance rate of 25% to 60%.46
As with the placebo responder, attempts have been made to characterize the potential drug defaulter on the basis of such factors as age, sex, education, race, and socioeconomic status. Although some correlations have been drawn (e.g., elderly patients are more apt to forget their medicine or to confuse one type of pill with another), many investigations have been either inconclusive or contradictory. The most important variables relate, not to the patient, but to the illness, the drug administered, the overall therapeutic regimen, and the doctor-patient relationship. Administration schedules are followed more faithfully by patients with life-threatening diseases than by patients with minor ailments. Even with serious illnesses such as essential hypertension, chronic infection, or hyperlipidemia, compliance is generally poor (approximately 50%) when the benefits of therapy are not superficially apparent.35 Drugs that produce unwanted side effects are especially likely to be discontinued. Deviations in self-administration tend to increase progressively with drugs that are taken long-term. Also, the more complex the therapeutic regimen in terms of doses and drugs, the higher the incidence of drug defaulting. The quality of the doctor-patient relationship is important in several respects. Patients who trust and respect their dentist or physician are more likely to take their prescribed medications. Effective communication further promotes compliance and reduces the possibility of a patient unilaterally terminating the drug if adverse effects occur. Measures that the clinician may use to enhance patient compliance are discussed in Chapter 55.
Drug interactions
The effect of a drug may be increased, decreased, or otherwise altered by the concurrent administration of another compound. Because agents routinely used in dental practice have been implicated in drug interactions, the topic is of considerable interest to the clinician and is addressed separately in Appendix 1.
ADVERSE DRUG REACTIONS
According to the Institute of Medicine, at least 1.5 million preventable adverse drug events occur annually in the United States.2 It has also been estimated that 5% to 17% of all patients hospitalized in the United States each year are admitted because of adverse reactions to drugs.5,28 Estimates of the annual cost of managing these reactions range from $3 billion to $7 billion.28 In addition, a survey of hospitalized patients between 1966-1996 revealed that 7% of hospitalized patients had a serious adverse drug reaction resulting in death, permanent disability, or prolonged care.22
Classification of Adverse Drug Reactions
Extension effects
Many drugs are used clinically in dosages that provide an intensity of effect that is submaximal. The reason for this conservatism is simple: increasing drug effects beyond a certain point may be dangerous. The anticoagulant warfarin is a typical example of a drug whose therapeutic action must be held in check to avoid serious toxicity. For the treatment of peripheral vascular thrombosis, warfarin is administered in doses that sufficiently increase the prothrombin time to yield an international normalized ratio (see Chapter 31) of 2 to 3. Warfarin could be given in larger amounts to inhibit clotting further, but the risk of spontaneous bleeding would be unacceptably high. Even with conventional therapy, hemorrhage—the toxic extension of warfarin’s anticoagulant effect—occurs in 2% to 4% of the patients treated. Inadvertent overmedication is one cause of warfarin toxicity; however, many additional factors influencing drug effects may also be involved, such as diet; heredity; gastrointestinal ulceration; genetic differences in drug metabolism; renal, hepatic, or cardiac insufficiency; drug interactions; and variable patient compliance. “Normal dose” has little meaning regarding warfarin because a therapeutic dose to one patient may represent an overdose to another.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


