Genetics in Pediatric Dentistry
Sven Kreiborg, Flemming Skovby, and Irma Thesleff
Most genetic disorders present in early childhood, some even in utero. The diagnosis and management of genetic disorders is therefore an important part of fetal medicine and pediatrics, including pediatric dentistry. Clinical genetics, which traditionally is responsible for genetic counseling, is an important collaborator.
Congenital malformations and syndromes
A congenital malformation can be isolated or be part of a syndrome with several malformations. Approximately 3% of newborns have one or more major malformations, defined as “major” by the patient’s need for medical intervention and the significance of the malformation for function and social acceptability. Subsequent to the finding of additional malformations, e.g., cardiac, during the first year of life, the frequency of major malformations increases to about 6%.
About 15% of newborns have minor malformations, an incidence somewhat depending on the accepted range of “normal.” Minor malformations should alert the clinician to significant disease, since 90% of children with three or more minor malformations also have a major malformation.
Dysmorphic features can be viewed as minor malformations. Some are inherited as autosomal dominant traits, as opposed to being part of a syndrome, and it is therefore important to evaluate a dysmorphic child on its familiar background. A malformation syndrome is characterized by manifestations in several organ systems from the same etiology, e.g., trisomy 21. Dysmorphologists are experts in syndrome identification.
Isolated malformations
When evaluating a congenital malformation in a child, it is crucial to clarify whether it is an isolated malformation or part of a syndrome with other, perhaps uncovered, organ manifestations. The prognosis for an isolated malformation is often favorable, and the recurrence risk is low. Conversely, the prognosis for malformation syndromes is often grave, especially if the central nervous system is involved, and the recurrence risk may be as high as 25–50%.
An isolated primary malformation may cause a sequence of structural defects. For example, a child with hypoplasia of the mandible (primary malformation) may also have cleft palate (secondary malformation or deformation), because the normal‐sized tongue did not allow for normal closure of the palate during fetal development, as seen in Robin sequence (Figure 25.1). Isolated malformations and their sequences include malformations, deformations, and disruptions.
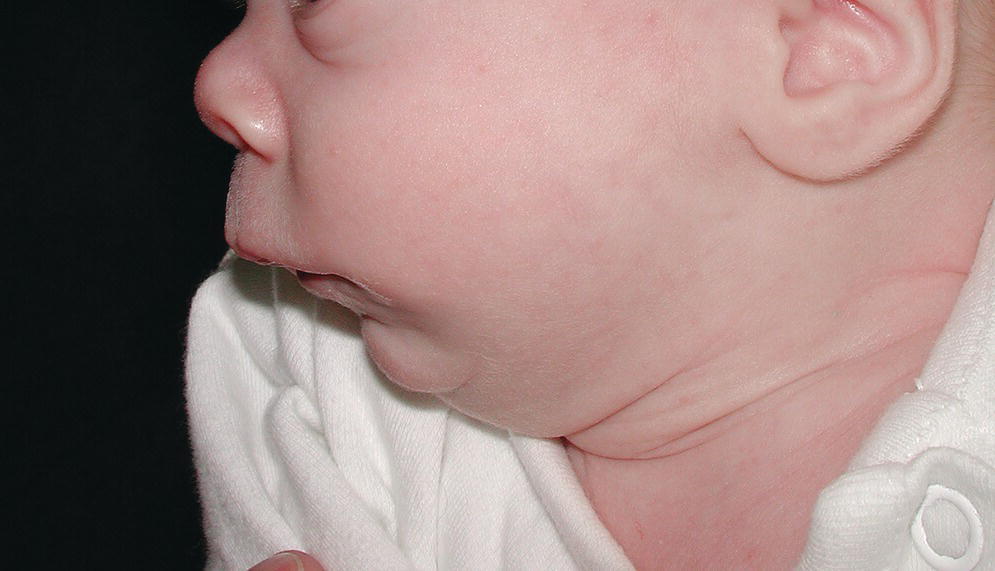
Figure 25.1 Infant with Robin sequence and cleft palate showing marked hypoplasia of the mandible.
Malformations are structural defects occurring during the formation of an organ or a tissue, e.g., congenital heart disease or micrognathia. The prognosis depends on the potential for treatment of the particular malformation, and it is often good, as for many congenital heart defects. The recurrence risk may be higher than in the general population, but it is usually less than 5% (see Table 25.1).
Table 25.1 Isolated malformations, deformations, and disruptions
| Type | Malformation Malformation sequence |
Deformation Deformation sequence |
Disruption Disruption sequence |
| Recurrence risk | <5% | Negligible | Negligible |
| Examples | Robin sequence, congenital heart defects, neural tube defect | Club foot and plagiocephaly due to intrauterine crowding | Facial clefts due to amniotic bands, constriction or amputation of extremities |
Deformations occur by external physical force during development of otherwise normal tissue or an organ. Deformations usually take place in late fetal life, e.g., dolichocephaly, torticollis, and hip luxation due to a breech presentation or clubfeet due to oligohydramnios. Most deformations have an excellent prognosis provided there is adequate physical therapy (see Table 25.1).
Disruptions are structural defects due to delay or interruption of normal development by vascular occlusion, infection, or mechanical force, e.g., an amniotic band. Disruptions have a very low recurrence risk.
Malformation syndromes
Although the genetic basis of syndromes is being unraveled with increasing speed, it still makes sense to categorize the well‐known malformation syndromes into the following groups based on different etiology: chromosomal disorders, microdeletion syndromes, monogenic disorders, teratogenic disorders, and disorders with unknown etiology. Many of the latter group turn out to have a genetic basis, obscuring the borders between the first three groups.
The etiology of a syndrome is important for treatment and prognosis, including genetic counseling. Knowing the natural history of a syndrome enables the clinician to target the follow‐up. Having established the diagnosis of a 22q11 deletion syndrome (or velo‐cardio‐facial syndrome), e.g., appropriate cardiac evaluation is mandatory, and carotid arterial variants close to the oral mucosa can be excluded before plastic surgery of the soft palate is undertaken for velo‐pharyngeal insufficiency.
Too much or too little chromosomal material is likely to result in congenital malformations, dysmorphic features, and psychomotor retardation. A number of deletions/duplications and monosomies/trisomies cause clinically recognizable syndromes, e.g., 22q11 deletion syndrome, cri du chat syndrome, Turner syndrome, and Down syndrome (trisomy 21).
Numerical or structural abnormalities of the sex chromosomes may cause gonadal insufficiency and either tall or short stature.
The size of most genes is between 1.5 and 2000 kb (1 kb = 103 nucleotide pairs). The resolution of a conventional banded karyotype is about 10 megabases (1 megabase = 106 nucleotide pairs), and a deletion may encompass many genes and not be revealed by this investigation. The current first‐choice cytogenetic investigation, a microarray or CGH analysis, has a 10 times greater resolution, and it will uncover a number of patients with microdeletion syndromes such as 22q11 deletion syndrome and Williams syndrome. (Array CGH analysis is a method analyzing the whole genome for quantitative chromosomal changes, deletions, and duplications.)
Disease‐causing mutations in single genes include substitutions, deletions, and duplications of one or more nucleotides. Mutations in the nuclear DNA follow mendelian inheritance, i.e., autosomal dominant, autosomal recessive, or X‐linked (see Box 25.1), while mutations in mitochondrial DNA can only be transmitted by females.
Many multiple malformation syndromes are monogenic, e.g., achondroplasia and Apert syndrome (see section “Syndromes with craniosynostosis”). DNA analysis clarifies an ever‐increasing number of monogenic disorders, increasing the possibilities for a precise diagnosis, genetic counseling, and prenatal diagnosis.
External forces can act on the developing fetus and cause dysmorphic traits and deformation sequences. Likewise, teratogens including toxic factors, medicines, alcohol, and narcotics cause such anomalies. In this context, the fetal alcohol syndrome is probably the most prevalent (see Figure 25.2).

Figure 25.2 A child with fetal alcohol syndrome. Note the small palpebral fissures, short nose, flat philtrum, and thin upper lip.
More than half of children with multiple malformation syndromes escape all current, routinely available diagnostic efforts. Significant improvement in diagnostic successes are expected with future use of next‐generation sequencing of patient DNA, either by whole exome sequencing or whole genome sequencing.
Craniofacial syndromes
About 1500 syndromes have been reported to present with craniofacial dysmorphology or malformation, and many of these syndromes also reveal dental anomalies (Hennekam et al. 2010). Among the syndromes with the most severe craniofacial manifestations are those associated with craniosynostosis and those affecting the pharyngeal arches. These patients present with complex craniofacial deformities that require a well‐planned, long‐term, staged, multidisciplinary approach to treatment. Such treatment is therefore centralized to major hospitals with a craniofacial team covering all the required medical and dental disciplines, including clinical genetics, pediatrics, neurosurgery, plastic surgery, oral and maxillofacial surgery, ophthalmology, ENT, orthodontics, and pediatric dentistry. In general, the degree of craniofacial phenotypic variability within each syndrome is remarkable, even within the same family. Therefore, the pediatric dentist has an important role in identifying the milder forms of craniofacial syndromes, especially those that affect the face, the jaws, and the dentition.
Syndromes with craniosynostosis
Craniosynostosis is premature fusion (most often prior to birth) of one or more cranial sutures of the calvarial vault. In addition, sutures of the cranial base, the orbits, and the maxillary complex may be involved. Some syndromes with craniosynostosis even have fusion of the synchondroses of the cranial base (e.g., Apert syndrome and Crouzon syndrome) [1].
Craniosynostosis is a serious abnormality of infancy and childhood requiring proper diagnosis and treatment (Cohen & MacLean 2000). It can be isolated or syndromic and proper diagnosis is essential for optimal patient care. Mutations in at least eight genes are unequivocally associated with mendelian forms of syndromic craniosynostosis: FGFR1, FGFR2, FGFR3, TWIST, EFNB1, MSX2, RAB23, and IL11RA, and these genes probably explain about 30% of the syndromic cases [2,3] (see Table 25.2). Heterozygosity for mutations in the fibroblast growth factor receptor genes FGFR1, FGFR2, and FGFR3 account for the majority of cases of syndromic craniosynostosis. It has been shown that activation of FGF signaling affects skeletal cell proliferation, adhesion, differentiation, and apoptosis, which may lead to synostosis of sutures [4]. Three of the most prevalent syndromes with craniosynostosis are presented below.
Table 25.2 Gene mutations causing craniosynostosis
| Syndrome | Phenotype | Causative gene | Mutations/transmission |
| Apert syndrome | Craniosynostosis Midface hypoplasia Syndactyly of hands and feet |
FGFR2 | Ser252Trp Pro253Arg Autosomal dominant |
| Carpenter syndrome | Craniosynostosis Syndactyly of hands and feet Postaxial polysyndactyly of feet |
RAB23 | >10 different mutations Autosomal recessive |
| Craniofrontonasal syndrome | Craniosynostosis Frontonasal dysplasia |
EFNB1 | >30 different mutations X‐linked |
| Craniosynostosis, Boston type | Craniosynostosis | MSX2 | Pro148His Autosomal dominant |
| Crouzon syndrome with acanthosis nigricans | Craniosynostosis Midface hypoplasia Ocular proptosis Acanthosis nigricans Jaw cementomas Vertebral anomalies |
FGFR3 | Ala391Glu Autosomal dominant |
| Crouzon syndrome | Craniosynostosis Midface hypoplasia Ocular proptosis |
FGFR2 | >30 different mutations Autosomal dominant |
| Jackson–Weiss syndrome | Craniosynostosis Broad great toes Tarsal/metatarsal coalitions |
FGFR2 | Ala344Gly Autosomal dominant |
| Kreiborg–Pakistani syndrome | Craniosynostosis Supernumerary teeth Delayed tooth eruption |
IL11RA | >5 mutations Autosomal recessive |
| Münke syndrome | Craniosynostosis (coronal suture) Hand anomalies (e.g., brachydactyly) |
FGFR3 | Pro250Arg Autosomal dominant |
| Pfeiffer syndrome | Craniosynostosis Midface hypoplasia Syndactyly of hands and feet Widening of thumbs and great toes |
FGFR2 FGFR1 |
>30 different mutations Autosomal dominant Pro252Arg Autosomal dominant |
| Saethre–Chotzen syndrome | Craniosynostosis Facial asymmetry Ptosis Soft tissue syndactyly 2nd and 3rd fingers and toes |
TWIST | >40 different mutations Autosomal dominant |
Münke syndrome is the most common syndromic form of craniosynostosis. It is an autosomal dominant condition caused by a point mutation, Pro250Arg, in the FGFR3 gene. The syndrome is characterized by unilateral or bilateral coronal synostosis and a variety of minor anomalies, e.g., brachydactyly and cone‐shaped epiphyses in the hands and feet, hearing loss, and, in some patients, developmental delay. It is estimated that about 30% of children with coronal synostosis and 6–8% of all craniosynostosis patients have this mutation. The craniofacial phenotype is highly variable. Several studies have reported that a phenotypically normal parent to a child with coronal synostosis and the Pro250Arg mutation in FGFR3 actually test positive for the same mutation.
Unilateral fusion of the coronal suture results in marked asymmetry of the craniofacial region. The frontal and parietal bones are flat and retruded on the ipsilateral side, and there is bossing of these bones on the contralateral side. The ipsilateral superior orbital rim is elevated and displaced posteriorly, and the palpebral fissure is wider and rounder on the ipsilateral side (see Figure 25.3). The nasal root deviates to the ipsilateral side, and the tip of the nose points to the contralateral side. The ear on the ipsilateral side is more anterior and higher than on the contralateral side. The orbital asymmetry may cause double vision with risk of unilateral blindness (amblyopia). If untreated, both the symmetry of the maxilla and the mandible become secondarily affected leading to facial asymmetry in all three planes of space and often with malocclusion in the transverse plane, i.e., unilateral crossbite [1].
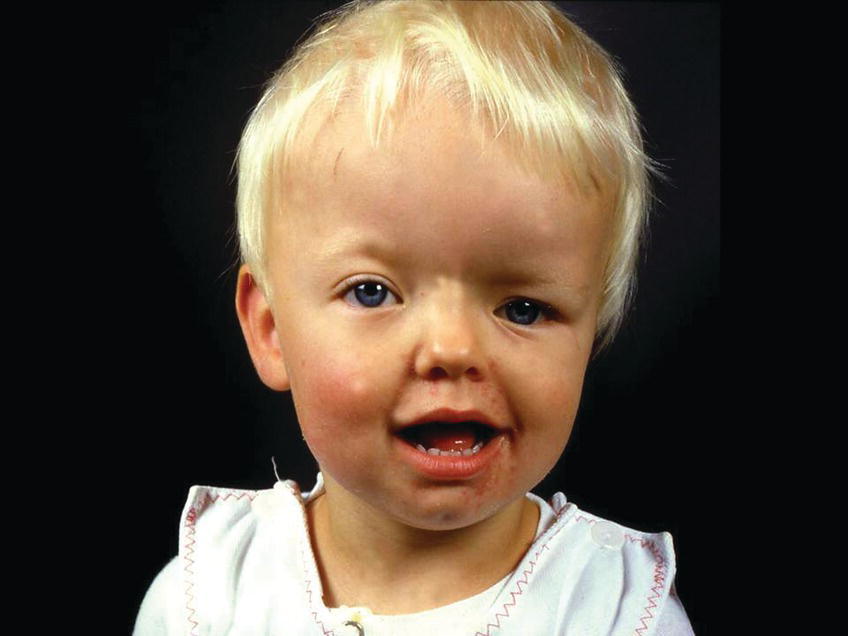
Figure 25.3 A 2‐year‐old girl with Münke syndrome and unicoronal synostosis. Note the marked craniofacial asymmetry.
Apert syndrome has an autosomal dominant inheritance, but most cases represent new mutations. New mutations are of paternal origin, and their risk of occurrence increases with increasing paternal age. Two different mutations in the FGFR2 gene in the linker region between immunoglobulin‐like loop II (IgII) and immunoglobulin‐like loop III (IgIII) (Ser252Trp and Pro253Arg) cause Apert syndrome. Activation of FGFR2 signaling in Apert syndrome probably results in premature osteoblast differentiation and fusion of craniofacial sutures and synchondroses. The syndrome is characterized by craniosynostosis, midface hypoplasia, and symmetric syndactyly of the hands and feet (see Figures 25.4 and 25.5).
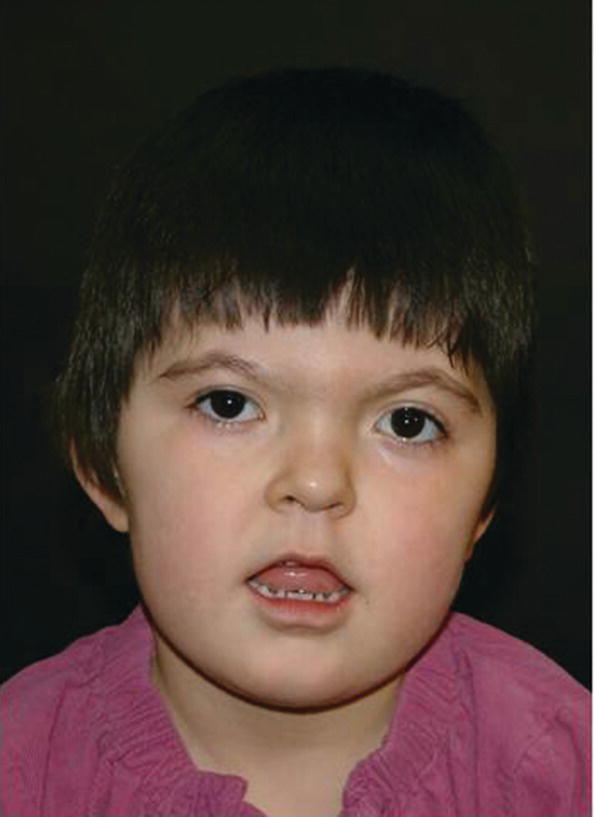
Figure 25.4 A 5‐year‐old girl with Apert syndrome. Note hypertelorism, short nose, trapezoidal shape of mouth, and midface hypoplasia.
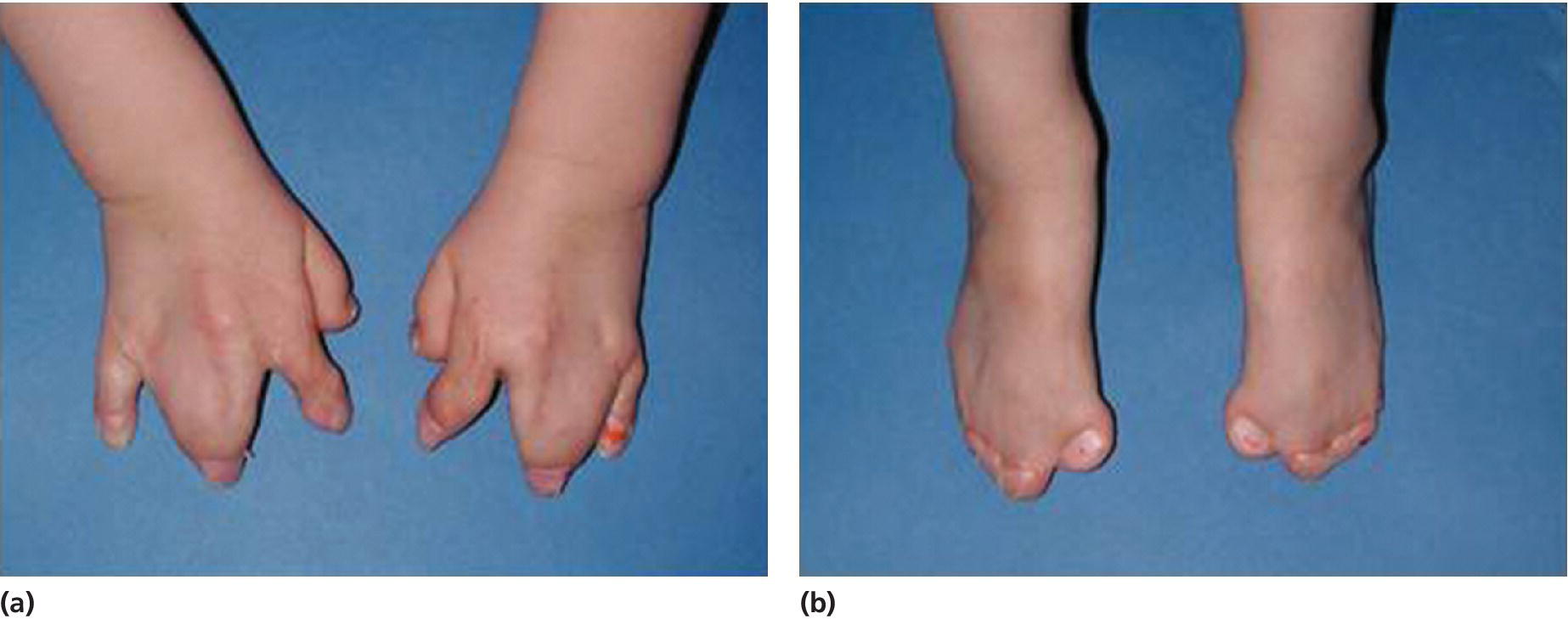
Figure 25.5 Symmetrical syndactyly of (a) hands and (b) feet in a 5‐year‐old girl with Apert syndrome. Early hand surgery was performed to release fingers 2 and 5.
At birth, the coronal sutures are invariably fused, whereas all other sutures are patent. In fact, most children have a wide midline defect in the calvaria extending from the glabellar area to the posterior fontanelle. This defect gradually ossifies without the formation of sutures. The skull is short, wide and, especially, tall (hyperbrachycephaly) (see Figure 25.6). Structural abnormalities of the CNS are common, and intelligence varies from normality to mental deficiency.
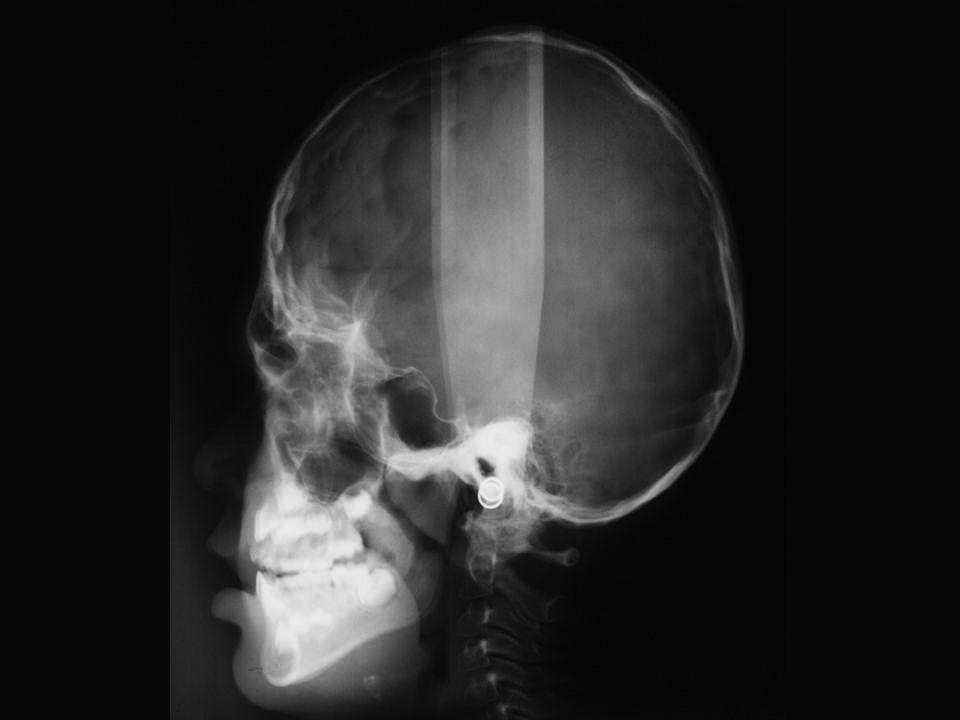
Figure 25.6 Skull X‐ray of a 5‐year‐old girl with Apert syndrome. Note craniosynostosis, hyperbrachycephaly, midface hypoplasia, and constriction of the nasopharyngeal airway.
The retrusion of the orbital rims and the maxillary hypoplasia lead to shallow orbits and ocular proptosis. The nose is short, the mouth has a trapezoidal shape, and most patients are mouth breathers because of the maxillary hypoplasia and the restricted nasopharyngeal airway. Clefting of the soft palate or uvula is common. The maxillary hypoplasia leads to severe crowding, delayed eruption and ectopic eruption of the teeth, and most often severe malocclusion with mandibular overjet, frontal open bite, and bilateral crossbite.
Crouzon syndrome has an autosomal dominant inheritance and is caused by mutations in FGFR2, and more than 30 different mutations in FGFR2 have been shown to be the cause. Around half the cases are caused by new mutations, and, similar to Apert syndrome, new mutations are of paternal origin, and their risk of occurrence increases with increasing paternal age. Activation of FGFR2 signaling in Crouzon syndrome probably results in premature osteoblast differentiation and fusion of craniofacial sutures and synchondroses. The syndrome includes craniosynostosis, hypertelorism, ocular proptosis, and midface hypoplasia (see Figure 25.7). Craniosynostosis is most often apparent at birth, but it may also develop during the first few years of life. Variable expression characterizes Crouzon syndrome even within the same family. The variable expression can most likely be explained by the fact that the pattern of fusion of sutures and synchondroses may vary both in location and in timing. Individuals with mild expression of the phenotype may remain undiagnosed although they have one of the known mutations in FGFR2.
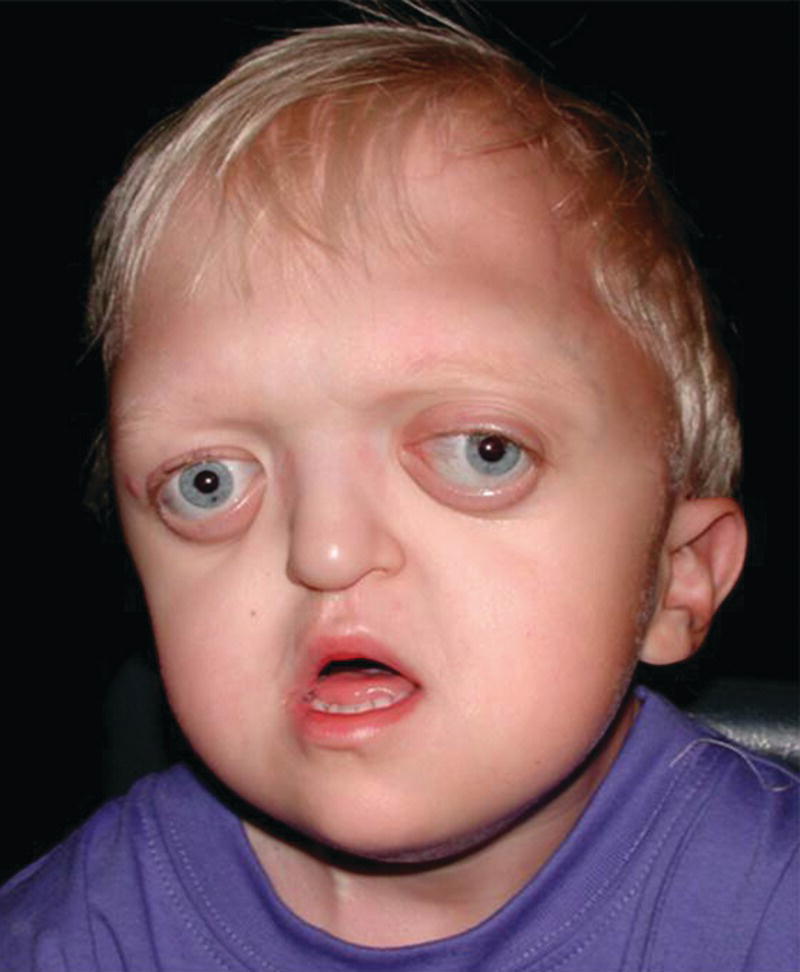
Figure 25.7 A 2‐year‐old boy with Crouzon syndrome. Note hypertelorism, ocular proptosis, and midface hypoplasia.
Skull shape is most often brachycephalic because of bicoronal synostosis, but dolichocephaly secondary to sagittal synostosis has also been reported. The craniosynostosis may lead to increased intracranial pressure and early neurosurgical treatment (within the first year of life) is most often required. The maxillary hypoplasia in all three planes of space contributes to the ocular proptosis, and it also may result in a compromised nasopharyngeal airway with risk of sleep apnea. In addition, maxillary hypoplasia leads to severe crowding, delayed eruption and ectopic eruption of the teeth, and most often to severe malocclusion with mandibular overjet, frontal open bite, and bilateral cross bite.
Crouzon syndrome with acanthosis nigricans is characterized by a complex craniosynostosis and a craniofacial phenotype similar to that of Crouzon syndrome (see Figure 25.8), but in addition the patients have skin thickening and hyperpigmentation especially at the flexures, and they may have cementomas of the jaws. It is caused by a specific mutation in the gene encoding FGFR3 (see Table 25.2).
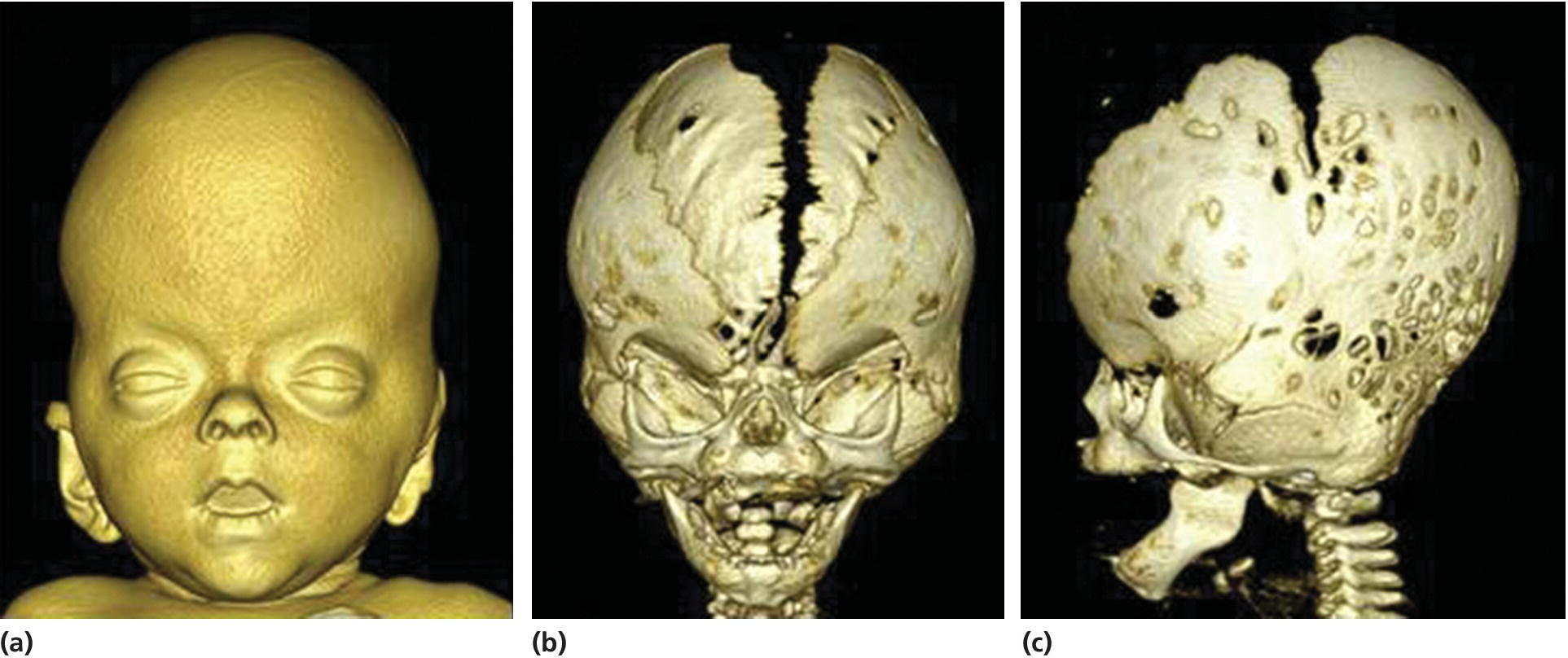
Figure 25.8 A 3D CT‐scanning of an infant with Crouzon syndrome with acanthosis nigricans. (a) Frontal view showing facial soft‐tissue morphology with hypertelorism, ocular proptosis, and midfacial hypoplasia. (b) Frontal view showing a wide calvarial midline defect. (c) Lateral view showing synostosis of the coronal and the lambdoid sutures and hyperbrachycephaly.
Syndromes affecting the pharyngeal arches
These congenital anomalies arise during embryogenesis from abnormal development of the first and second pharyngeal arches and their derivatives. The anomalies manifest at birth with eye and ear defects combined with maxillary, zygomatic and mandibular hypoplasia, and frequently with cleft palate. The syndromes can be subdivided into mandibulofacial dysostoses and acrofacial dysostoses, which also have limb defects. The spectrum covers several different conditions, e.g., hemifacial microsomia, Goldenhar syndrome, Treacher Collins syndrome, and Nager syndrome. The best understood is Treacher Collins syndrome.
Treacher Collins syndrome is also known as mandibulofacial dysostosis. It is primarily associated with autosomal dominant mutations in the TCOF1 gene, which is located on chromosome 5. More than 200 different mutations have been documented throughout the gene including deletions, insertions, splicing, and missense and nonsense zygote mutations. Variable expression characterizes Treacher Collins syndrome even within the same family, and it is not uncommon for mildly affected individuals to be diagnosed retrospectively after the birth of a severely affected child [5]. Animal models of Treacher Collins syndrome have elucidated the pathogenesis of the disorder. Most craniofacial manifestations of the disorder seem to be caused by problems in the development of cells of the neural crest. Thus, it has been suggested that there is a deficit in the number of migrating neural crest cells by as much as 25%. Therefore, the general craniofacial hypoplasia observed in individuals with Treacher Collins syndrome probably arises from a deficiency in the number of migrating neural crest cells from which the majority of the cranial bones and cartilages are derived [5].
The phenotype is characterized by downslanting palpebral fissures, colobomas of the lower eyelids, and absence of the medial third of the lower eyelashes; there is hypoplasia of the facial bones, especially of the mandible and the zygomatic complex, but the maxilla is also involved, and cleft palate is common (see Figure 25.9). There is often an inferolateral orbital cleft, and the zygomatic arch is often missing. In addition, there is microtia, atresia of the external auditory canals, and abnormalities of the ossicles of the middle ear with hearing loss. Hypoplasia of the mandible with marked retrognathia and a decreased posterior mandibular height have in severe cases resulted in perinatal death because of respiratory problems. The airway problems seem to be worsened by progressive decrease of the cranial base angle [6,7]. Even in milder cases, respiratory problems and feeding problems may hamper somatic development in early childhood. The mandibular hypoplasia most often leads to severe Class II malocclusion with extreme maxillary overjet, frontal open bite, and severe crowding of the teeth.
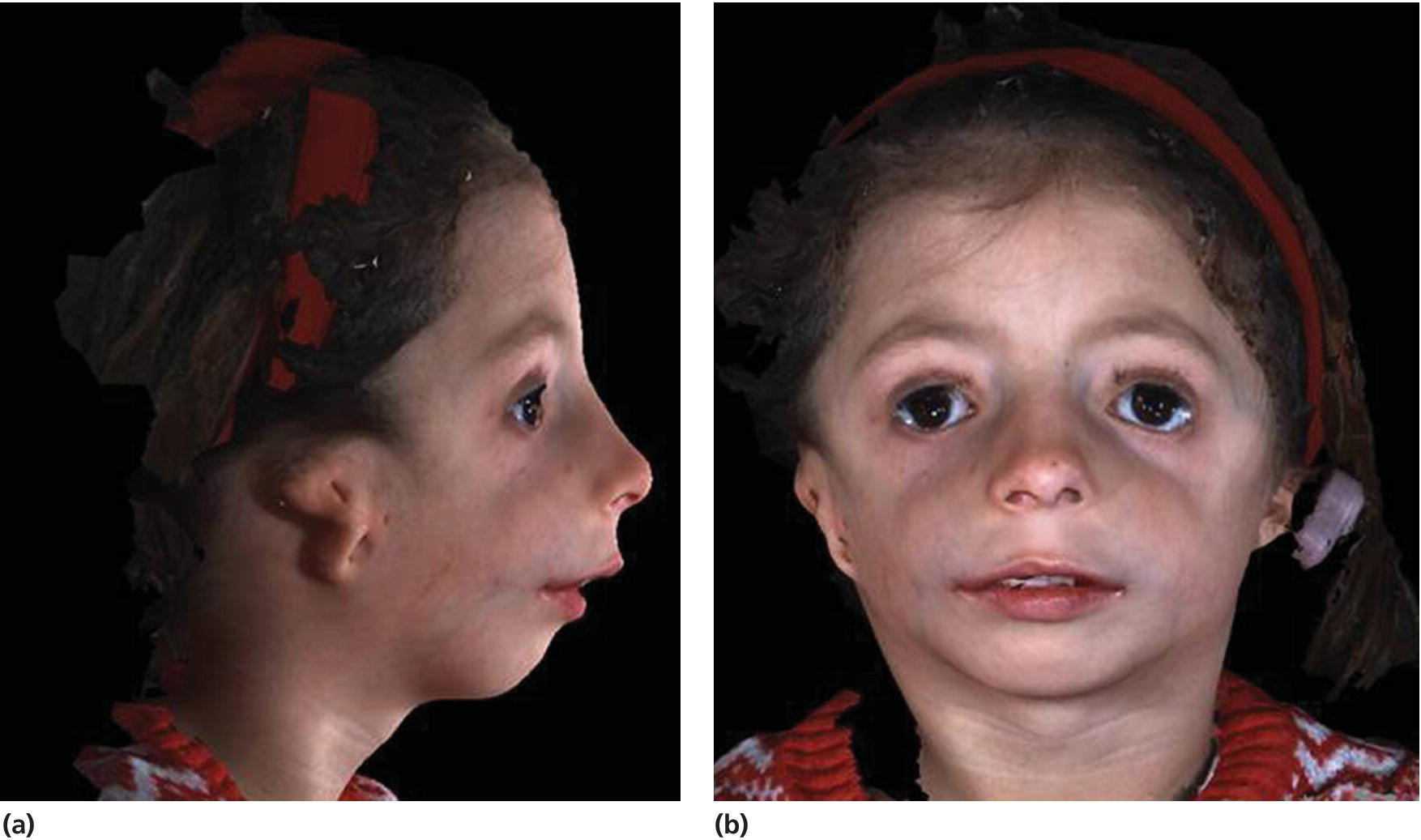
Figure 25.9 A 5‐year‐old girl with Treacher Collins syndrome. Note the downslanting palpebral fissures, colobomas of the lower eyelids, mandibular hypoplasia, and microtia.
Dental anomalies
Tooth development is under strict genetic control and, apart from some mineralization disturbances which are commonly caused by environmental factors, dental anomalies result from gene mutations. It is noteworthy that most mutations that have so far been identified as causing abnormalities in tooth number and/or shape affect molecules of the signaling networks regulating early tooth morphogenesis (see Chapter 4). In contrast, most of the currently known mutations that cause inherited defects in dentin and enamel are in genes encoding major components of their respective extracellular matrices.
Congenitally missing teeth
The gene defects causing the most common form of hypodontia, affecting one or a few incisors and/or premolars, have not yet been identified, but mutations causing isolated (nonsyndromic) and syndromic forms of oligodontia (severe hypodontia, defined in Box 4.3) have been found in several genes (see Table 25.3). WNT10A has come up as the gene most commonly associated with oligodontia. So far, about half of the mutations causing isolated (nonsyndromic) oligodontia have been found in the WNT10A gene [8]. In addition, WNT10A mutations are frequent causes of various types of ectodermal dysplasias (syndromes affecting several ectodermal organs including teeth, hair, glands, and nails), in particular when associated with oligodontia. It has been suggested that WNT10A mutation analysis might become an important diagnostic test in ectodermal dysplasias with oligodontia as well as in isolated oligodontia [9,10]. WNT10A encodes a signal molecule expressed in dental epithelium during tooth development.
Table 25.3 Gene mutations causing aberrations in number and shape of teeth
| Syndrome | Phenotype | Causative gene | Type of molecule |
| Oligodontia +/– ectodermal dysplasia | Severe hypodontia Defects in hairs and nails |
WNT10A | Signal molecule |
| Oligodontia | Severe hypodontia | PAX9 | Transcription factor |
| Oligodontia | Severe hypodontia Cleft palate (occasionally) |
MSX1 | Transcription factor |
| Oligodontia–colorectal cancer syndrome | Hypodontia of permanent teeth Colorectal cancer |
AXIN2 | Wnt inhibitor |
| Rieger syndrome | Oligodontia Eye and umbilical defects |
PITX2 (RIEG) | Transcription factor |
| Hypohidrotic ectodermal dysplasia | Oligodontia Small peg‐shaped teeth Sparse hair Deficiency of salivary and sweat glands |
EDA/Ectodysplasin EDAR EDARADD |
Signal molecule EDA receptor EDA mediator |
| EEC syndrome (Ectrodactyly‐ectodermal dysplasia‐clefting syndrome) | Ectodermal dysplasia Ectrodactyly Cleft lip/palate |
P63 | Transcription factor |
| CLPED (Cleft lip/palate‐ectodermal dysplasia syndrome) | Ectodermal dysplasia Cleft lip/palate |
PVRL1 | Cell adhesion molecule (Nectin‐1) |
| Cleidocranial dysplasia | Supernumerary teeth Impaired eruption Deficient bone formation |
RUNX2 | Transcription factor |
| Kreiborg–Pakistani syndrome | Supernumerary teeth Impaired eruption Craniosynostosis |
IL11RA | Interleukin receptor |
| Adenomatous polyposis coli FAP (Gardner syndrome) |
Supernumerary teeth Odontomas Impacted teeth |
APC | Wnt inhibitor |
Autosomal dominant isolated oligodontia can be caused by mutations in the transcription factor genes MSX1 and PAX9 [11,12]. Patients with PAX9 mutations have no other congenital defects whereas patients with MSX1 mutations may have cleft palate. MSX1 and PAX9 have central functions in the early dental mesenchyme where they mediate epithelial–mesenchymal signaling. Mutations in AXIN2, an inhibitor of Wnt signaling, cause a rare form of oligodontia affecting only permanent teeth. These patients may develop colorectal cancer later in life [13].
Most of the hypodontia genes that have been identified are associated with syndromes affecting several other organs besides the teeth. That the development of different organs is disturbed by mutations in one gene is not surprising, taking into account the fact that similar mechanisms and the same genes regulate the development of many organs. The causative gene in Rieger syndrome is PITX2 [14], encoding a transcription factor that is considered as the best marker of tooth formation and dental identity. Oligodontia is most commonly associated with various forms of ectodermal dysplasias and several causative genes have been identified in addition to WNT10A. In hypohidrotic ectodermal dysplasia (HED) oligodontia is severe (sometimes all teeth are missing) and the rest of the teeth are small and peg‐shaped [15,16]. This syndrome is usually caused by the loss of function of the signal molecule ectodysplasin (EDA) belonging to the tumor necrosis factor family, or other molecules participating in the mediation of EDA signaling, e.g., its receptor EDAR [17]. Neonatal injections of EDA protein can rescue the phenotype in the mouse and dog models of HED (caused by EDA mutations) and, intriguingly, this may present a cure for this form of human HED as well [18]. Ectodermal dysplasia may be associated with cleft lip and palate, and mutations in the transcription factor P63 and the cell adhesion molecule nectin‐1 have been identified as causes of such syndromes (see Table 25.3).
Hyperdontia (supernumerary teeth)
Compared to hypodontia, hyperdontia, or the formation of supernumerary teeth, is quite rare, and the genetic background has been clarified in few conditions (see Table 25.3). Cleidocranial dysplasia is an autosomal dominant syndrome mainly affecting bone development (hypoplastic clavicles, open fontanelles, short stature) [19]. The patients have multiple extra teeth, which develop successively from the permanent (secondary) teeth and form a partial third dentition [20]. In addition, about 30% of patients have supernumerary molars. The eruption of teeth is impaired most likely because of defective bone remodeling [21]. The causative gene is RUNX2, a transcription factor that is a master regulator of osteoblasts, the bone‐forming cells. Experiments in mouse embryos have shown that in addition to bone, the gene is expressed in dental mesenchyme during the bud and bell stages of tooth development, and that it is critically involved in signaling interactions between the epithelium and mesenchyme.
Supernumerary teeth have been reported as part of the Kreiborg–Pakistani craniosynostosis syndrome caused by mutation in the IL11RA gene [2]. This gene encodes the receptor of interleukin 11, a signal molecule expressed in mesenchymal cells of the dental papilla as well as around the tooth. The eruption of teeth is delayed in this syndrome apparently due to the defect in bone remodeling. Supernumerary teeth together with odontomas and impacted teeth occur in 10–20% of patients with familial adenomatosis coli (FAP). The causative gene is APC, which is a modulator of Wnt signaling. Interestingly, the stimulation of Wnt signaling in dental epithelium of transgenic mice results in massive production of supernumerary teeth which develop successively from previously formed teeth [22]. Hence, the Wnt signal pathway likely is a key regulator of tooth initiation and replacement.
Aberrations in the structure of dental hard tissues
Amelogenesis imperfecta refers to hereditary defects in enamel formation. In cases where the enamel defects are the only symptoms, most mutations have been identified in genes that encode specific enamel matrix proteins, such as amelogenin and enamelin (see Table 25.4). In addition, mutations in genes encoding enzymes required for the degradation of amelogenin and enamelin, including MMP20 and KLK4, cause amelogenesis imperfecta. Enamel defects occur also as traits in several syndromes, mostly in association with skin diseases and inborn errors of metabolism. Epidermolysis bullosa, a hereditary blistering skin disease, is sometimes associated with defective enamel. The blisters result from failure of the adhesion of epithelium to stromal tissue and are due to mutations in basement membrane components such as laminins. The defective enamel in these cases likely results from compromised adhesion of ameloblasts to enamel matrix. Genetic variation in KRT75 gene encoding the intracellular protein keratin 75 has been shown to disturb the structure of enamel leading to increased risk of dental caries [23]. Keratin 75 is expressed in ameloblasts but is best known as an epithelial hair keratin and thus its mutations also cause abnormalities such as curly hair.
Table 25.4 Genes with mutations associated with defects in dental hard tissues
| Syndrome | Phenotype | Causative gene | Type of molecule |
| Amelogenesis imperfecta | Thin hypoplastic and pitted enamel | AMELX/Amelogenin | Major enamel matrix protein |
| Amelogenesis imperfecta | Thin hypoplastic and pitted enamel | ENAM/Enamelin | Enamel matrix protein |
| Amelogenesis imperfecta | Hypomineralized, soft and pigmented enamel | MMP20/Enamelysin | Matrix metalloproteinase |
| Amelogenesis imperfecta | Hypomineralized, soft and pigmented enamel | KLK4/Kallikrein4 | Matrix metalloproteinase |
| Amelogenesis imperfecta | Deficient enamel | KRT75/Keratin75 | Intermediate filament protein |
| Epidermolysis bullosa | Hypoplastic enamel Fragile skin Blisters |
LAMA 3, LAMB3, LAMC2/Laminins | Basement membrane components |
| Tricho‐dento‐osseous syndrome (TDO) | Enamel defects Taurodontism Hair and bone defects |
DLX3 | Transcription factor |
| Dentinogenesis imperfecta | Dentin dysplasia | DSPP/Dentin sialophosphoprotein | Dentin matrix protein |
| Osteogenesis and dentinogenesis imperfect | Dentin dysplasia Discolored teeth Brittle bones |
COL1A1/2/Type1 collagen | Extracellular matrix protein |
| Hypophosphatasia | Lack of cementum | ALPL/Alkaline phosphatase | Hydrolase enzyme |
Dentinogenesis imperfecta and dentin dysplasia are severe dentin defects that affect both crown and root dentin. Mutations in the dentin matrix component dentin sialophosphoprotein (DSPP) cause dentinogenesis imperfecta and dentin dysplasia. The major extracellular matrix molecule of dentin is type 1 collagen, and mutations in the COL1 genes cause dentinogenesis imperfecta. As type 1 collagen is also the main matrix component of bone, this type of dentinogenesis imperfecta is associated with osteogenesis imperfecta, a disabling disease of brittle bones (see Box 25.2).
VIDEdental - Online dental courses


