CHAPTER 25 Drugs Used in Treating Heart Failure
Heart failure is characterized by a decreased ability of the heart to pump blood in adequate amounts. In addition to changes in the heart, multiple adaptive mechanisms occur, which provide targets for drug therapy. The severity of heart failure is often rated according to the New York Heart Association (NYHA) classification, ranging from class I, in which signs of heart failure occur only at higher exercise levels, to class IV, in which signs of heart failure occur at rest. In addition to drug therapy, other strategies for treating heart failure include auxiliary mechanical pumps (ventricular assist devices)4 and ventricular pacing (resynchronization).
CARDIAC MUSCLE CONTRACTION AND HEART FAILURE
Drugs such as the β-adrenergic receptor agonists increase cardiac contractility by increasing intracellular cyclic 3′,5′-adenosine monophosphate (cAMP), which enhances Ca++ influx and accelerates uptake of Ca++ by the SR, ultimately making more Ca++ available for contraction.12 The latter effect results from the phosphorylation of phospholamban, a protein associated with the Ca++ pump of the SR. The effect of Ca++ on troponin may also be enhanced by cAMP.
In heart failure, the heart is unable to maintain the requisite cardiac output. The mechanics underlying this failure are incompletely understood. The ability of the SR to participate in the trafficking of Ca++ seems to be hindered.21 The Na+-Ca++ exchange sites seem to be increased in heart failure, leading to a decrease in intracellular Ca++. There are likely to be multiple biochemical defects in heart failure, however.
According to Starling’s law of the heart, cardiac output, or, more precisely, the ventricular stroke volume, increases as ventricular filling pressure increases. Stated simply, the heart pumps whatever is supplied to it by way of venous return, maintaining a near-optimal heart size. As ventricular end-diastolic pressure increases, ventricular stroke work and stroke volume increase. Normal heart function is within well-defined limits and is described by a single curve (Figure 25-1).
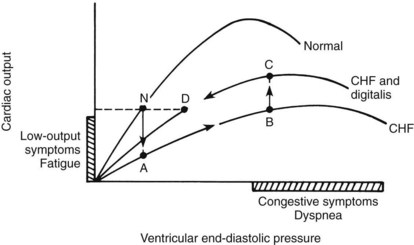
When cardiac contractility is reduced in heart failure, three mechanisms are available by which the heart can compensate for the defect: (1) an increase in ventricular end-diastolic pressure, which enhances cardiac output (Frank-Starling preload mechanism); (2) an increase in number of contractile units (hypertrophy); and (3) use of chronotropic and inotropic reserves of the heart through reflex mechanisms (sympathetic activity). If these mechanisms are sufficient to produce normal cardiac output, the heart failure is said to be compensated. In this condition, a new ventricular function curve is generated (see Figure 25-1). For any given ventricular end-diastolic pressure, however, ventricular stroke work, stroke volume, and cardiac output are lower in the failing heart than in the normal heart. Consequently, the heart enlarges to maintain cardiac output, and heart rate increases to help compensate for poor cardiac function.
Figure 25-2 shows some important adaptive mechanisms that result from heart failure. These changes, including an increase in sympathetic discharge, can compensate for the heart failure. If these and other responses are insufficient, however, the heart failure becomes uncompensated. Adaptive mechanisms also include an increase in production of angiotensin II, leading to remodeling of the heart over time.10 Remodeling results in cardiac hypertrophy, and several cellular changes, which, although they tend to compensate for heart failure, may hasten the course of the disease. This cardiac remodeling in heart failure has a parallel in hypertension, in which vascular smooth muscle slowly undergoes hypertrophy and hyperplasia.
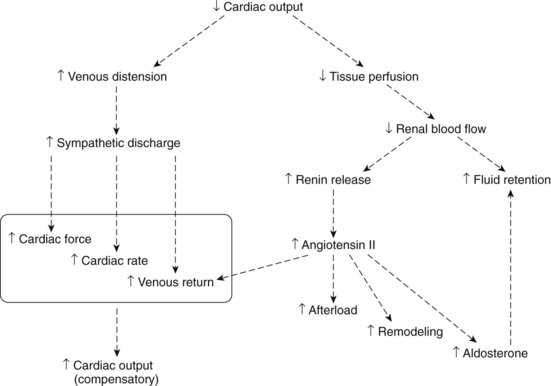
As a result of the complexity of changes in heart failure, there are numerous processes that can be targeted by drugs: neurohumoral events, vascular dynamics, fluid volume, the sympathetic nervous system, and contractility of the heart.10 The pharmacologic features of angiotensin-converting enzyme (ACE) inhibitors and other vasodilators, diuretics, β blockers, and catecholamines are discussed elsewhere in this book. They are also discussed in this chapter in relation to the treatment of heart failure. The cardiac glycosides are not extensively discussed elsewhere in the book and so are discussed more fully in this chapter than other drugs used for heart failure.
DRUGS USED IN THE TREATMENT OF CHRONIC HEART FAILURE
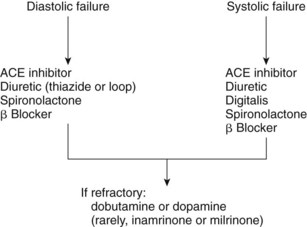
Table 25-1 lists the drugs used to treat heart failure and their mechanisms of action. The drugs are often used in combination. Heart failure can be classified as diastolic or systolic failure. In diastolic failure, the heart has inadequate distention and inadequate filling capabilities. Contraction as measured by the ejection function may be normal. This type of heart failure is often seen in patients with hypertension. Systolic heart failure is a deficiency in contractility with a low ejection fraction.
| DRUG OR DRUG CLASS | MECHANISM(S) |
|---|---|
| Long-Term Treatment | |
| Thiazide and loop diuretics | Reduce fluid volume (reduce preload and afterload) |
| ACE inhibitors | Reduce effect of angiotensin II, prevent remodeling |
| Angiotensin II receptor blockers | Reduce effect of angiotensin II, prevent remodeling |
| β Blockers* | Reduce sympathetic effect, prevent remodeling, prevent arrhythmias |
| Aldosterone antagonists | Inhibit effect of aldosterone |
| Digoxin | Direct cardiotonic effect |
| Isosorbide dinitrate-hydralazine | Reduce afterload and preload |
| Short-Term Treatment† | |
| Dobutamine | Direct cardiotonic effect |
| Dopamine | Direct cardiotonic effect |
| Nesiritide | Reduces preload and afterload |
| Phosphodiesterase III inhibitors | Reduce preload and afterload; direct cardiotonic effect |
| Nitroglycerin | Reduces preload and afterload |
| Nitroprusside | Reduces preload and afterload |
ACE, Angiotensin-converting enzyme.
* Carvedilol is a β blocker that also blocks α1 adrenoceptors.
† Usually only for a few days.
The primary drugs used to treat chronic heart failure are ACE inhibitors, angiotensin II receptor blockers, thiazide and loop diuretics, β-adrenergic receptor blockers, aldosterone antagonists, digoxin, and directly acting vasodilators. For short-term acute treatment, certain catecholamines, nesiritide, other vasodilators, and the phosphodiesterase III inhibitors have special application. Figure 25-3 shows the drugs used in each type of heart failure.4,7
Aldosterone Antagonists
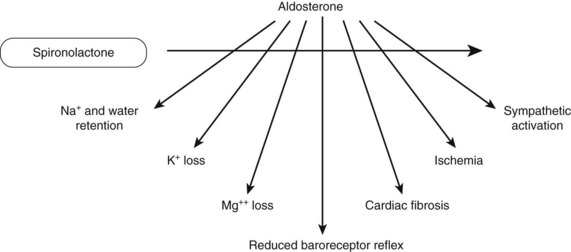
The action of spironolactone as a diuretic is caused by its antagonism of aldosterone at the convoluted tubule of the kidney. Antagonism of aldosterone leads to several other effects that are beneficial in patients with heart failure. These are shown in Figure 25-4. K+-sparing actions help in preventing hypokalemia. Reducing Mg++ loss seems to reduce ventricular arrhythmias in patients with heart failure. Because aldosterone can inhibit norepinephrine uptake (uptake 2), spironolactone prevents the enhanced sympathetic activation from aldosterone.18 Spironolactone also blocks the inhibition of the baroreceptor reflex seen with aldosterone. A consequence of inhibiting the baroreceptor reflex is the lack of parasympathetic nerve response.13 The latter response is important in counteracting the adverse effects of sympathetic stimulation, such as arrhythmias and cardiac ischemia. Myocardial fibrosis is also inhibited by spironolactone.
Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers
A major effect of ACE inhibitors is to reduce afterload and preload. These drugs improve symptoms in patients with heart failure. In addition, the progressive deterioration of the heart is slowed with ACE inhibitors. ACE inhibitors control remodeling that occurs with chronic heart failure. Remodeling results from growth of the myocardial cell and myocardial fibrosis. This probable cardioprotective effect provides added support for the use of ACE inhibitors in heart failure. Inhibiting the production of angiotensin II or blocking its receptor reduces aldosterone secretion, which reduces Na+ and water retention. Reduction of aldosterone release by ACE inhibitors also reduces sympathetic discharge, as described in Chapter 28 (see Figure 25-4).
Evidence also suggests that ACE inhibitors stimulate the proliferation of capillaries in the coronary circulation, increasing blood flow. It is unknown whether this action of ACE inhibitors occurs as a result of the decrease in angiotensin II, an increase in bradykinin, or both. Most likely, the ability of ACE inhibitors to reduce blood pressure in hypertensive patients also contributes to a cardioprotective effect. Early treatment of congestive heart disease with an ACE inhibitor could be a major factor in slowing the progress of the disease and in relieving symptoms. Enalapril is one of the first drugs whose administration was associated with an increased survival time in patients with heart failure.15 These drugs also are useful in combination with other drugs, including digoxin and diuretics, in treating heart failure.
Angiotensin II antagonists, such as losartan and valsartan, are additional candidates for treating heart failure.7,10 They share the benefits of ACE inhibitors, while avoiding such side effects as angioedema and persistent coughing. The pharmacologic features of angiotensin II antagonists and ACE inhibitors are discussed in detail in Chapter 28.
β-Adrenergic Receptor Antagonists
The use of β-adrenoceptor blockers to reduce the adverse cardiac effects of heightened sympathetic discharge that occur as heart failure progresses is a strategy used in the long-term treatment of heart failure.20 β Blockers reduce the work of the heart, reduce renin secretion, prevent remodeling of the heart, act as antiarrhythmic drugs, and reduce the downregulation of β1-adrenergic receptors in heart failure. All of these effects are beneficial in heart failure. Their use is consistent with a neurohumoral component of heart failure. Bisoprolol, betaxolol, and metoprolol are selective β1-adrenergic receptor blockers that are used in treating heart failure.
Directly Acting Vasodilators
Vasodilators such as nitrates and hydralazine reduce the load on the heart, improve tissue perfusion in heart failure, and increase survival rates in these patients. Vasodilators are usually administered in combination with other drugs, such as inotropic agents or ACE inhibitors. Disadvantages of nitrates and hydralazine include the indirect enhancement of sympathetic discharge and activation of the renin-angiotensin pathway. Added interest in vasodilator therapy resulted from evidence supporting the clinical efficacy of two vasodilators in combination: isosorbide dinitrate and hydralazine. The fixed-dose therapy was found to be effective in patients of African descent with class III and class IV heart failure.3,17 This drug combination may be particularly effective in this group of patients, who may be less responsive to drugs such as ACE inhibitors. The isosorbide dinitrate–hydralazine combination may also be useful as added therapy in resistant cases or in patients who cannot tolerate other heart failure medications. The pharmacologic characteristics of nitrates and hydralazine are discussed in Chapters 26 and 28, respectively.
DIGOXIN
The first detailed scientific study of digitalis on record was made by Sir William Withering19 of Shropshire, England, in 1785. In his treatise, “An Account of the Foxglove, and Some of Its Medical Uses; With Practical Remarks on Dropsy, and Other Diseases,” Withering detailed clinical uses for the leaf of the Digitalis purpurea (purple foxglove) plant and described its effects on the heart. His recognition of the potential usefulness of digitalis plant derivatives was occasioned by their extensive use in local folk medicines. Withering ascribed the beneficial effects of digitalis in treating dropsy to a direct diuretic effect, although he was aware of beneficial effects on the heart as well. He also detailed many toxic effects of the plant.
Chemistry and Classification
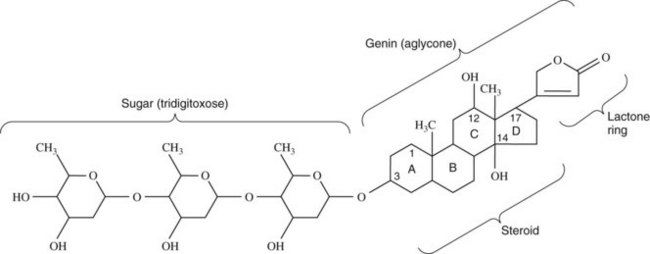
The term digitalis is often used interchangeably with the term cardiac glycoside. Both terms refer to many compounds, naturally occurring or semisynthetic, that have similar cardiotonic effects. Only one such compound, digoxin, is commonly used clinically in the United States today. Another agent, digitoxin, is available in Canada and elsewhere. The structure of digoxin is shown in Figure 25-5. The molecule is composed of a steroid ring structure. Other distinguishing molecular characteristics include an α,β-unsaturated lactone ring, and a carbohydrate moiety in glycosidic linkage at C3. The presence of a sugar in glycosidic linkage accounts for the name glycoside. A steroid plus lactone lacking the sugar group is generically called a genin or aglycone. The genin of digoxin is digoxigenin.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


