Metabolic disorders
Key Points
• The metabolic syndrome is increasingly recognized and potentially fatal
• Inborn metabolic errors are rare but may be life-threatening
Metabolic Syndrome
Metabolic syndrome (metabolic syndrome X, insulin resistance syndrome, dysmetabolic syndrome, hypertriglyceridaemic waist, obesity syndrome, Reaven syndrome) is the name for a group of risk factors that increase the risk for ischaemic heart disease (IHD), diabetes and stroke (Fig. 23.1). The metabolic syndrome is diagnosed when at least three of the IHD risk factors listed in Table 23.1 are present. Whether the syndrome, which affects possibly 25% of the US population, is a specific syndrome, and nothing more than the sum of its parts, is controversial.
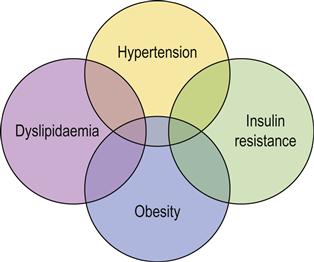
Table 23.1
< ?comst?>
| Features | Detaila |
| Hypertension | Blood pressure≥130/85 mmHg |
| Hyperglycaemia | Fasting blood glucose≥5.6 mmol/L |
| Dyslipidaemia | Triglycerides≥150 mg/dL |
| High-density lipoprotein (HDL)<50 mg/dL for women and<40 mg/dL for men | |
| Abdominal obesity | Large waistline:≥35 inches for women and≥40 inches for men |
< ?comen?>< ?comst1?>

< ?comst1?>
< ?comen1?>
In general, a person with metabolic syndrome is twice as likely to develop IHD and five times as likely to develop diabetes as someone without it. The probability of developing metabolic syndrome is also closely linked to a lack of physical activity and the fact of being overweight/obese. Other causes include insulin resistance, ethnicity (often Asian), family history, older age and other factors (Box 23.1). Associated diseases and signs may be raised uric acid levels, hepatic steatosis, haemochromatosis and acanthosis nigricans. Metabolic syndrome may be associated with inflammatory periodontal disease.
Treatment of metabolic syndrome is to increase physical activity but to reduce weight, unhealthy diet, low-density lipoprotein cholesterol and high blood pressure; diabetes should also be controlled (Chs 5 and 6).
Inborn Errors of Metabolism
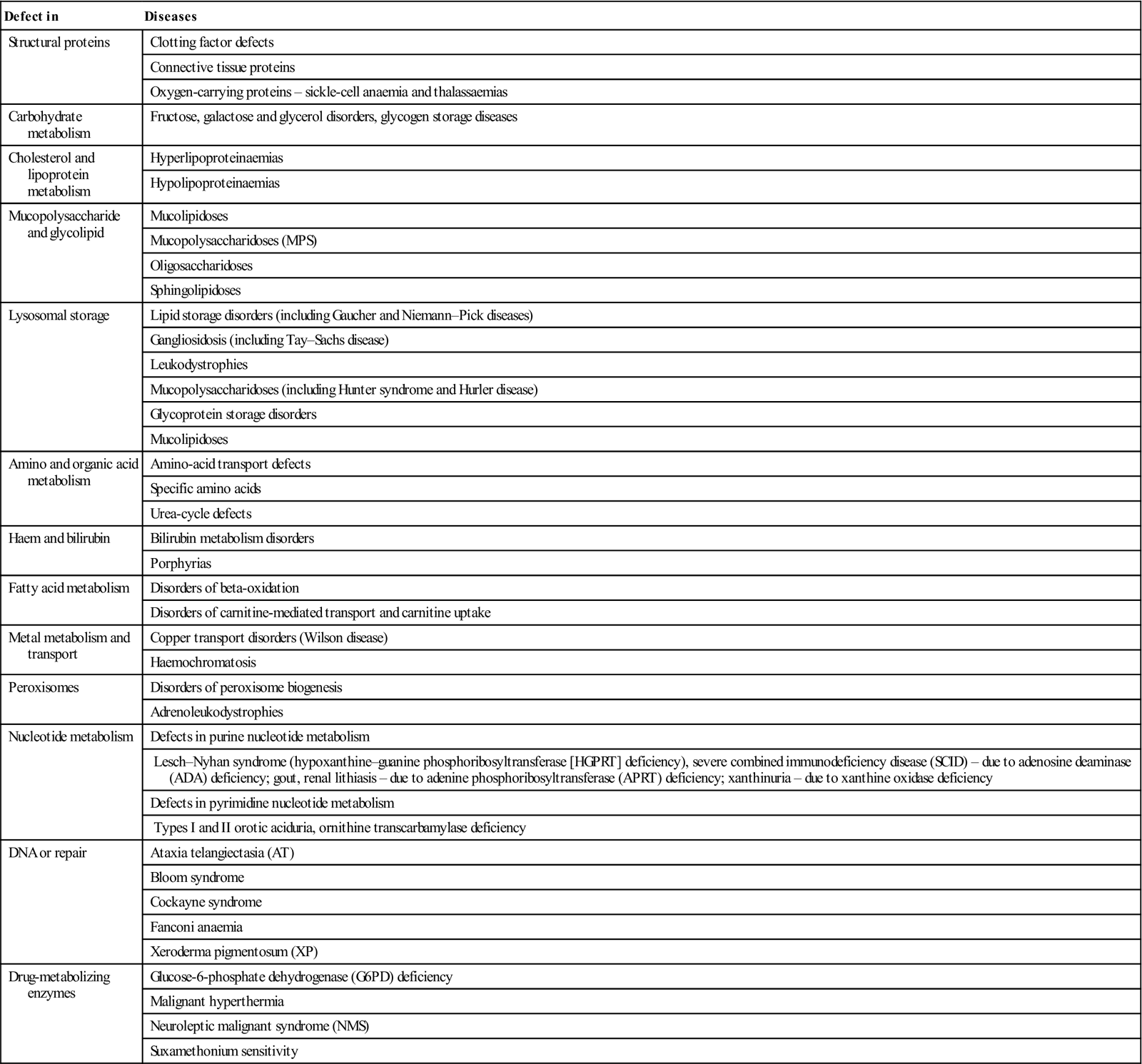
There are a vast number of rare metabolic disorders that result from inherited enzyme defects; some have a high prevalence in certain ethnic groups (Table 23.2). Most arise from recessive traits and thus manifest only when both parents are heterozygous carriers. They affect 1 in 4 of the offspring of heterozygous parents, often as a result of first-cousin marriage.
Table 23.2
< ?comst?>
| Defect in | Diseases |
| Structural proteins | Clotting factor defects |
| Connective tissue proteins | |
| Oxygen-carrying proteins – sickle-cell anaemia and thalassaemias | |
| Carbohydrate metabolism | Fructose, galactose and glycerol disorders, glycogen storage diseases |
| Cholesterol and lipoprotein metabolism | Hyperlipoproteinaemias |
| Hypolipoproteinaemias | |
| Mucopolysaccharide and glycolipid | Mucolipidoses |
| Mucopolysaccharidoses (MPS) | |
| Oligosaccharidoses | |
| Sphingolipidoses | |
| Lysosomal storage | Lipid storage disorders (including Gaucher and Niemann–Pick diseases) |
| Gangliosidosis (including Tay–Sachs disease) | |
| Leukodystrophies | |
| Mucopolysaccharidoses (including Hunter syndrome and Hurler disease) | |
| Glycoprotein storage disorders | |
| Mucolipidoses | |
| Amino and organic acid metabolism | Amino-acid transport defects |
| Specific amino acids | |
| Urea-cycle defects | |
| Haem and bilirubin | Bilirubin metabolism disorders |
| Porphyrias | |
| Fatty acid metabolism | Disorders of beta-oxidation |
| Disorders of carnitine-mediated transport and carnitine uptake | |
| Metal metabolism and transport | Copper transport disorders (Wilson disease) |
| Haemochromatosis | |
| Peroxisomes | Disorders of peroxisome biogenesis |
| Adrenoleukodystrophies | |
| Nucleotide metabolism | Defects in purine nucleotide metabolism |
| Lesch–Nyhan syndrome (hypoxanthine–guanine phosphoribosyltransferase [HGPRT] deficiency), severe combined immunodeficiency disease (SCID) – due to adenosine deaminase (ADA) deficiency; gout, renal lithiasis – due to adenine phosphoribosyltransferase (APRT) deficiency; xanthinuria – due to xanthine oxidase deficiency | |
| Defects in pyrimidine nucleotide metabolism | |
| Types I and II orotic aciduria, ornithine transcarbamylase deficiency | |
| DNA or repair | Ataxia telangiectasia (AT) |
| Bloom syndrome | |
| Cockayne syndrome | |
| Fanconi anaemia | |
| Xeroderma pigmentosum (XP) | |
| Drug-metabolizing enzymes | Glucose-6-phosphate dehydrogenase (G6PD) deficiency |
| Malignant hyperthermia | |
| Neuroleptic malignant syndrome (NMS) | |
| Suxamethonium sensitivity |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Some inborn errors of metabolism, especially suxamethonium sensitivity, malignant hyperpyrexia, hyperlipoproteinaemias and porphyria, can seriously complicate management if general anaesthesia (GA) is required (see Table 23.2 and Appendix 23.1). Many others are associated with learning impairment or neurological disease. Some others can also be relevant occasionally, such as haemochromatosis, homocystinuria and glycogen storage diseases. A few (e.g. hereditary pentosuria) are innocuous (see Appendix 23.1). Few inborn errors of metabolism will be encountered in primary care, but they are more common in paediatric or special care clinics.
Defects in Carbohydrate Metabolism
Gaucher disease
Gaucher disease is a rare lysosomal storage disease, common in Ashkenazi Jews; it is caused by glucocerebrosidase deficiency. Manifesting with hepatosplenomegaly, skeletal involvement (including honeycomb-like mandibular radiolucencies), anaemia and thrombocytopenia, it may present challenges with bleeding and – should the patient be splenectomized – a liability to infection.
Glycogen storage diseases
General aspects
Glycogen storage diseases (GSD) are rare disorders due to inherited defects in degradative enzymes that mobilize glycogen to glucose, so that it accumulates and hypoglycaemia results.
Clinical features
Glycogen accumulates in liver and muscle, resulting in hepatomegaly, muscle pain, weakness, respiratory muscle weakness and cardiac failure. Hypoglycaemia is common, as are infections.
Dental aspects
Enlarged tongue, periodontal breakdown and masticatory muscle pain may be complaints. Patients with GSD type 1b have a bleeding tendency that can be corrected preoperatively by 24–48 hours of glucose infusion. Local anaesthesia (LA) appears to have no contraindications. Conscious sedation (CS) may be given if required. GA requires expert attention in hospital.
Defects in fructose metabolism
Hereditary fructose intolerance (aldolase B deficiency) and fructosuria – hepatic fructokinase deficiency – discourage the patient from ingesting sugar (sucrose) and are therefore associated with low dental caries rates.
Defects in Cholesterol and Lipoprotein Metabolism
Hyperlipoproteinaemia (hyperlipidaemia; hypercholesterolaemia)
General aspects
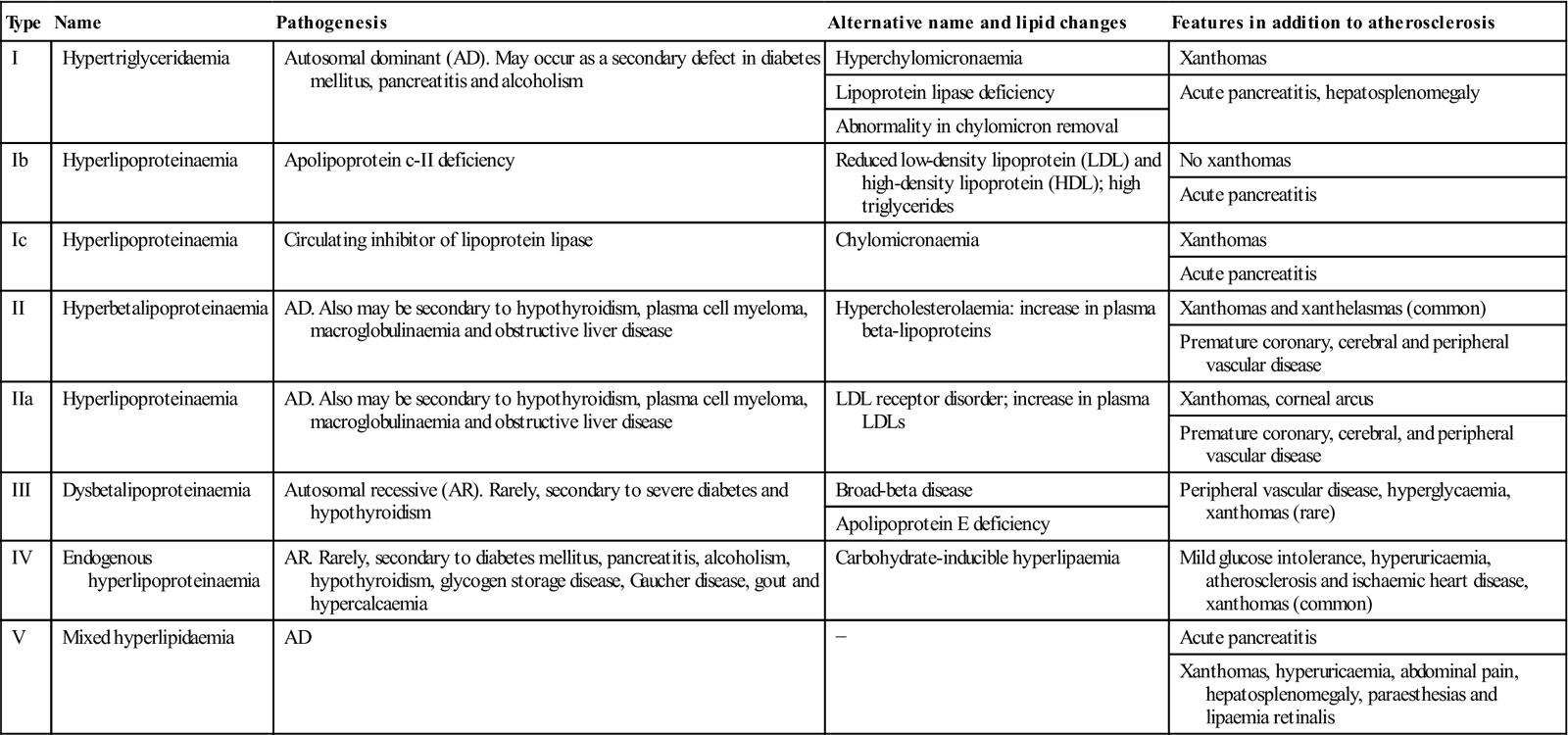
Cholesterol is found in some hormones, as well as in every cell, and thus is an essential element. However, high cholesterol (hypercholesterolaemia), which is directly related to hyperlipoproteinaemia (Table 23.3), predisposes to atherosclerosis (Ch. 5).
Table 23.3
Primary hyperlipoproteinaemias
< ?comst?>
| Type | Name | Pathogenesis | Alternative name and lipid changes | Features in addition to atherosclerosis |
| I | Hypertriglyceridaemia | Autosomal dominant (AD). May occur as a secondary defect in diabetes mellitus, pancreatitis and alcoholism | Hyperchylomicronaemia | Xanthomas |
| Lipoprotein lipase deficiency | Acute pancreatitis, hepatosplenomegaly | |||
| Abnormality in chylomicron removal | ||||
| Ib | Hyperlipoproteinaemia | Apolipoprotein c-II deficiency | Reduced low-density lipoprotein (LDL) and high-density lipoprotein (HDL); high triglycerides | No xanthomas |
| Acute pancreatitis | ||||
| Ic | Hyperlipoproteinaemia | Circulating inhibitor of lipoprotein lipase | Chylomicronaemia | Xanthomas |
| Acute pancreatitis | ||||
| II | Hyperbetalipoproteinaemia | AD. Also may be secondary to hypothyroidism, plasma cell myeloma, macroglobulinaemia and obstructive liver disease | Hypercholesterolaemia: increase in plasma beta-lipoproteins | Xanthomas and xanthelasmas (common) |
| Premature coronary, cerebral and peripheral vascular disease | ||||
| IIa | Hyperlipoproteinaemia | AD. Also may be secondary to hypothyroidism, plasma cell myeloma, macroglobulinaemia and obstructive liver disease | LDL receptor disorder; increase in plasma LDLs | Xanthomas, corneal arcus |
| Premature coronary, cerebral, and peripheral vascular disease | ||||
| III | Dysbetalipoproteinaemia | Autosomal recessive (AR). Rarely, secondary to severe diabetes and hypothyroidism | Broad-beta disease | Peripheral vascular disease, hyperglycaemia, xanthomas (rare) |
| Apolipoprotein E deficiency | ||||
| IV | Endogenous hyperlipoproteinaemia | AR. Rarely, secondary to diabetes mellitus, pancreatitis, alcoholism, hypothyroidism, glycogen storage disease, Gaucher disease, gout and hypercalcaemia | Carbohydrate-inducible hyperlipaemia | Mild glucose intolerance, hyperuricaemia, atherosclerosis and ischaemic heart disease, xanthomas (common) |
| V | Mixed hyperlipidaemia | AD | − | Acute pancreatitis |
| Xanthomas, hyperuricaemia, abdominal pain, hepatosplenomegaly, paraesthesias and lipaemia retinalis |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
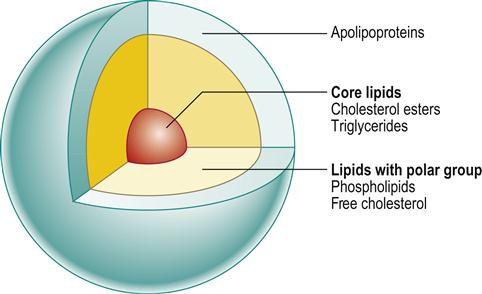
The lipoproteins consist of lipids (insoluble in plasma), phospholipids and apolipoproteins (apoproteins). Based on density and electrophoretic mobility, the lipoproteins are divided into four groups:
■ Chylomicrons are large particles carrying mainly triglyceride to the liver, where lipoprotein lipase releases free fatty acids (Fig. 23.2).
Hypercholesterolaemia can be due to abnormalities in lipoprotein levels related to diet, or secondary to obesity, diabetes, hypothyroidism, nephrotic syndrome, chronic renal failure, liver disease, alcoholism, smoking (lowers HDL) and use of oral contraceptives. In primary (familial) hyperlipidaemia (FH), genetic factors play a role, with defects, for example, in the LDL receptor (LDLR), apolipoprotein B-100 (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes.
Clinical features
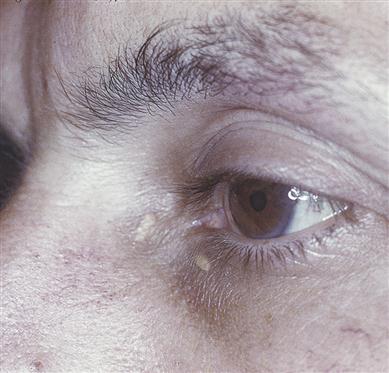
Gout, arthralgias, arthritis and premature IHD may result. Yellowish cutaneous plaques called xanthomas, in soft tissue, tendons and subperiosteal and intraosseous locations (xanthelasmas on the eyelids; Fig. 23.3), are associated with accelerated IHD. Prominent xanthomas on elbows and knees, especially with yellow to orange discolouration of palmar creases, are unique to type II hyperlipoproteinaemia.
General management
Around 75% of FH is undiagnosed, so screening for LDLR, APOB and PCSK9 genes may help. Dietary intervention and lipid-lowering drugs (particularly statins – HMG-CoA reductase inhibitors) are essential to treatment (Ch 5). Ezetimibe is an option for primary hypercholesterolaemia.
Dental aspects
LA is the safest method of pain control. CS can be given if required. GA may be a risk because of IHD.
Facial xanthomas may help recognition of these potentially dangerous diseases. Types IV and V hyperlipoproteinaemias may be complicated by sicca syndrome.
Hypolipoproteinaemias
Pulpal calcifications, unusual odontomes and orange deposits in the tonsils may be found in HDL deficiency (Tangier disease).
Errors in Amino Acid and Organic Acid Metabolism
Homocysteine (Hcy), a sulphur amino acid, is an intermediate product in conversion of methionine to cysteine; it participates in the re-methylation pathway, enabling maintenance of adequate cellular levels of methionine, or is catabolized by trans-sulphuration. Several hereditary defects in the enzymes involved and acquired deficiencies in the vitamin cofactors of these enzymes can cause hyperhomocysteinaemia.
Hyperhomocysteinaemia
An increased homocysteine level is associated with a higher risk of strokes. Carotid stenosis appears to have a graded response to increased levels of homocysteine; increased carotid plaque thickness has been associated with high homocysteine and low B12 levels. Homocysteine may also be a risk factor for Alzheimer disease. Dietary folate and B12 supplementation may normalize homocysteine levels and modify the risk of vascular disease.
Homocystinuria
Homocystinuria is a disorder of methionine metabolism, leading to an abnormal accumulation of homocysteine and its metabolites (homocystine, homocysteine–cysteine complex, and others) in blood and urine. Homocystinuria is an autosomal recessively inherited defect in the trans-sulphuration pathway (homocystinuria I) or methylation pathway (homocystinuria II and III).
Affected patients are tall with a Marfanoid appearance, joint hypermobility, ectopia lentis, and visual and mental deterioration. Blood vessel abnormalities, together with excessive platelet adhesiveness, predispose to thromboembolism, both spontaneously and postoperatively. Pyridoxine is the treatment. During and after surgery, aggressive hydration and prophylaxis for deep vein thrombosis (DVT) are recommended.
Defects in Non–Amino-Acid Nitrogen Metabolism
The porphyrias
General aspects
Porphyrias are rare inborn errors in enzymes involved in haem metabolism, which result in the accumulation of porphyrins, the intermediate compounds in haem(oglobin) synthesis (Fig. 23.4). Classification is according to the principal site of enzyme defect: namely, liver (hepatic porphyrias) or erythrocytes (erythropoietic porphyrias; Table 23.4).
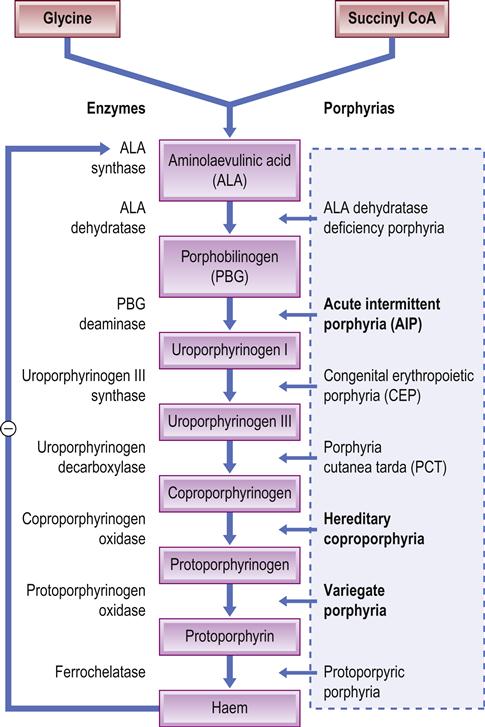
Table 23.4
< ?comst?>
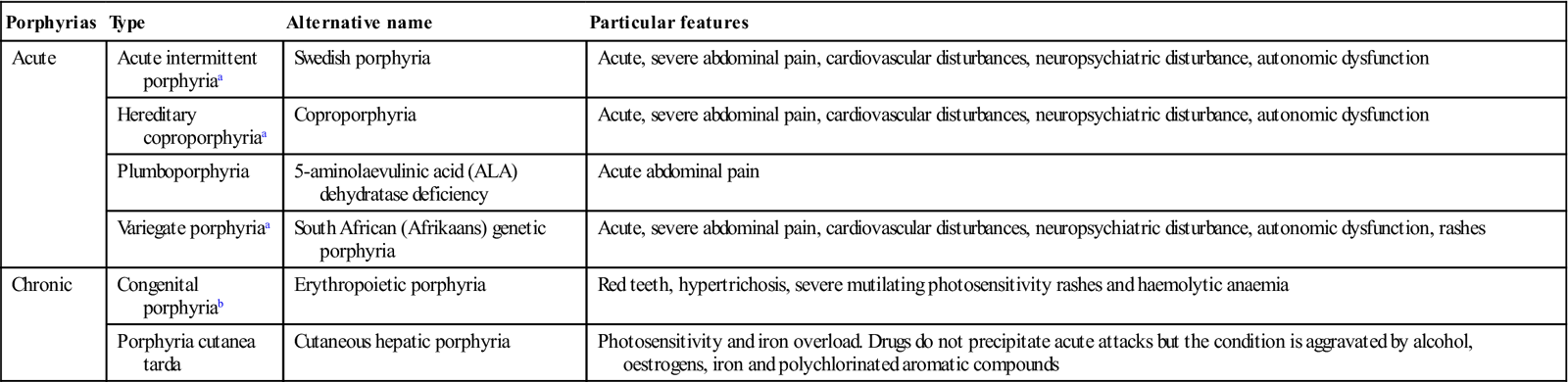
| Porphyrias | Type | Alternative name | Particular features |
| Acute | Acute intermittent porphyriaa | Swedish porphyria | Acute, severe abdominal pain, cardiovascular disturbances, neuropsychiatric disturbance, autonomic dysfunction |
| Hereditary coproporphyriaa | Coproporphyria | Acute, severe abdominal pain, cardiovascular disturbances, neuropsychiatric disturbance, autonomic dysfunction | |
| Plumboporphyria | 5-aminolaevulinic acid (ALA) dehydratase deficiency | Acute abdominal pain | |
| Variegate porphyriaa | South African (Afrikaans) genetic porphyria | Acute, severe abdominal pain, cardiovascular disturbances, neuropsychiatric disturbance, autonomic dysfunction, rashes | |
| Chronic | Congenital porphyriab | Erythropoietic porphyria | Red teeth, hypertrichosis, severe mutilating photosensitivity rashes and haemolytic anaemia |
| Porphyria cutanea tarda | Cutaneous hepatic porphyria | Photosensitivity and iron overload. Drugs do not precipitate acute attacks but the condition is aggravated by alcohol, oestrogens, iron and polychlorinated aromatic compounds |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Most people with porphyrias enjoy entirely normal health. However, three porphyrias are significant clinically, since drugs precipitate acute illness. Patients may develop potentially fatal acute attacks of cardiovascular (hypertension and tachycardia) and gastrointestinal disturbances, accompanied by neuropsychiatric disturbance (porphyria in King George III of England probably explains his ‘madness’). These porphyrias are variegate porphyria, the commonest, found mainly in South Africans of Afrikaans descent; acute intermittent porphyria, seen less frequently but affecting all population groups; and hereditary coproporphyria, which is rare and may cause patients to appear normal, apart from severe photosensitivity rashes.
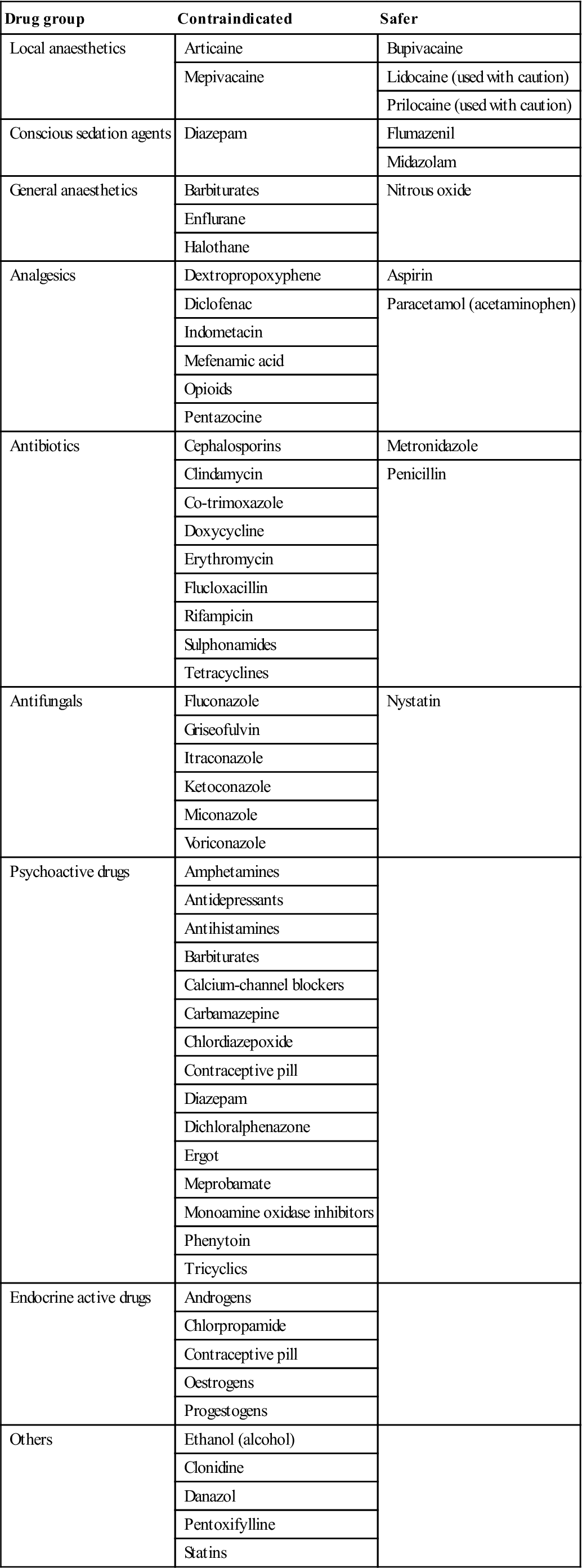
Most of the drugs that have been incriminated in attacks are enzyme inducers that also increase hepatic 5-aminolaevulinic acid (ALA) synthetase levels. Assessing drug safety in porphyria is an inexact science; for many drugs no such information is available, and for some others the data are of doubtful validity and often conflicting (Table 23.5). A wide array of other factors can cause exacerbations, including fasting, alcohol, infections and pregnancy.
Table 23.5
< ?comst?>
| Drug group | Contraindicated | Safer |
| Local anaesthetics | Articaine | Bupivacaine |
| Mepivacaine | Lidocaine (used with caution) | |
| Prilocaine (used with caution) | ||
| Conscious sedation agents | Diazepam | Flumazenil |
| Midazolam | ||
| General anaesthetics | Barbiturates | Nitrous oxide |
| Enflurane | ||
| Halothane | ||
| Analgesics | Dextropropoxyphene | Aspirin |
| Diclofenac | Paracetamol (acetaminophen) | |
| Indometacin | ||
| Mefenamic acid | ||
| Opioids | ||
| Pentazocine | ||
| Antibiotics | Cephalosporins | Metronidazole |
| Clindamycin | Penicillin | |
| Co-trimoxazole | ||
| Doxycycline | ||
| Erythromycin | ||
| Flucloxacillin | ||
| Rifampicin | ||
| Sulphonamides | ||
| Tetracyclines | ||
| Antifungals | Fluconazole | Nystatin |
| Griseofulvin | ||
| Itraconazole | ||
| Ketoconazole | ||
| Miconazole | ||
| Voriconazole | ||
| Psychoactive drugs | Amphetamines | |
| Antidepressants | ||
| Antihistamines | ||
| Barbiturates | ||
| Calcium-channel blockers | ||
| Carbamazepine | ||
| Chlordiazepoxide | ||
| Contraceptive pill | ||
| Diazepam | ||
| Dichloralphenazone | ||
| Ergot | ||
| Meprobamate | ||
| Monoamine oxidase inhibitors | ||
| Phenytoin | ||
| Tricyclics | ||
| Endocrine active drugs | Androgens | |
| Chlorpropamide | ||
| Contraceptive pill | ||
| Oestrogens | ||
| Progestogens | ||
| Others | Ethanol (alcohol) | |
| Clonidine | ||
| Danazol | ||
| Pentoxifylline | ||
| Statins |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Clinical features of attacks
Often a single dose of a drug of this type can precipitate an acute episode, but in some patients, repeated doses are necessary to provoke a reaction. Attacks are accompanied by cardiovascular disturbance (tachycardia and hypertension, sometimes followed by postural hypotension). Respiratory embarrassment or major convulsions and neuropsychiatric disturbance (typically a peripheral sensory and motor neuropathy, but sometimes agitation, mania, depression, hallucinations and schizophrenic-like behaviour) may occur. Autonomic dysfunction may cause profuse sweating, pallor and pyrexia. Severe hyponatraem/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


