Chapter 22
Disorders of Red Blood Cells
Disorders of the red blood cells (RBCs), which in large part consist of the anemias, are of clinical importance in dental practice for several reasons. First, the dentist serves an important role in detecting patients with anemia through history, clinical examination, and results of screening laboratory tests. These screening procedures should lead to early referral to a physician and the establishment of the diagnosis. Clinical recognition of anemia can significantly affect morbidity and mortality risks, because anemia often occurs as an underlying condition that requires attention and medical treatment. Also of note, anemia is an independent risk factor for adverse cardiovascular outcomes (i.e., acute myocardial infarction and death) in a variety of patient populations (as defined by chronic kidney disease, acute coronary syndrome, or old age, for example).< ?xml:namespace prefix = "mbp" />
Anemia
Definition
Anemia, which is defined as a reduction in the oxygen-carrying capacity of the blood, usually is associated with a decreased number of circulating RBCs or an abnormality in the Hb contained within the RBCs. Anemia is not a disease but rather a symptom complex that may result from one of three underlying causes: (1) decreased production of RBCs (iron deficiency, pernicious anemia, folate deficiency), (2) blood loss, or (3) increased rate of destruction of circulating RBCs (hypersplenism, autoimmune destruction).
Erythropoiesis
About 1% of the circulating erythrocyte mass is generated by the bone marrow each day. Precursors of RBCs are reticulocytes, which account for 1% of the total RBC count. The normal RBC is about 33% hemoglobin by volume. Hemoglobin (Hb), the oxygen-carrying molecule of erythrocytes, consists of two pairs of globin chains (i.e., α plus β, δ, or γ) that form a shell around four oxygen-binding heme groups. Healthy adults have about 95% HbA (α2β2) and small amounts of HbA2 (α2δ2) and HbF (α2γ2). Genes on chromosome 16 encode α globin chains; β chains are encoded on chromosome 11.
Epidemiology
It is estimated that about 3.4 million Americans have anemia.
Etiology
Anemia has numerous causes (
| Classification by RBC Size and Shape | Cause |
|---|---|
| Microcytic (MCV ≤ 80 fL |
|
| Iron deficiency anemia | Decreased production of RBCs |
| Thalassemias | Defective hemoglobin synthesis |
| Lead poisoning | Inhibition of hemoglobin synthesis |
| Normocytic (MCV 80-100 fL |
|
| Increased destruction of RBCs | |
| Aplastic anemia | Decreased production of RBCs |
| Renal failure | Decreased production of RBCs |
| Anemia of chronic disease | Decreased production of RBCs |
| Macrocytic (MCV > 100 fL |
|
| Pernicious anemia | Decreased production of RBCs |
| Folate deficiency | Decreased production of RBCs |
| Hypothyroidism | Decreased production of RBCs |
fL, Femtoliter; MCV, Mean corpuscular volume; RBC, Red blood cell.
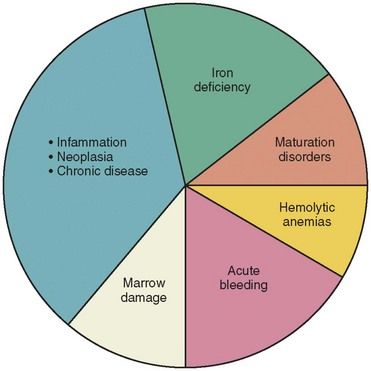
Types of Anemia
Iron Deficiency Anemia
Iron deficiency anemia is a microcytic anemia (
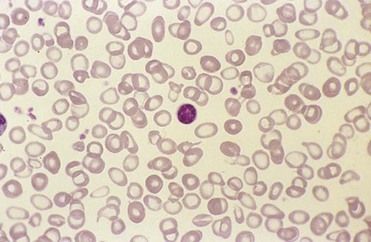
FIGURE 22-2 Microcytic anemia associated with iron deficiency. Peripheral blood smear shows red blood cells (RBCs) that are small and have marked hypochromic central pallor.
In women, menstruation and pregnancy contribute to the development of iron deficiency anemia. The repeated loss of blood associated with menses can lead to depletion of iron, resulting in a mild state of anemia. During pregnancy, the expectant mother experiences an increased demand for additional iron and vitamins to support the growth of her fetus, and unless sufficient amounts of these nutrients have been provided in some form, she may become anemic. Approximately 20% of pregnant women have iron deficiency anemia.
By contrast, mild anemia in men usually indicates the presence of a serious underlying medical problem (e.g., gastrointestinal bleeding, malignancy). Under normal physiologic conditions, men lose little iron, and because iron can be stored for months, iron deficiency anemia is rare in men. Therefore, any man who is found to be anemic should be promptly referred for medical evaluation.
Folate Deficiency Anemia and Pernicious Anemia
Vitamin B12 (cobalamin) and folic acid are needed for RBC formation and growth within bone marrow. Vitamin B12 is a cofactor in methionine-associated enzymatic reactions required of protein synthesis and thus in the maturation of RBCs. Folate is needed for enzymatic reactions required for the synthesis of purines and pyrimidines of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) and thus for the synthesis of proteins. A deficiency in daily intake (as in chronic alcoholism) or absorption (due to celiac disease or tropical sprue) of these vitamins can result in anemia.
Folate is found in fruits and leafy vegetables. It is not stored in the body in large amounts, so a continual dietary supply is needed. Its absorption and metabolism are interfered with by alcohol consumption and certain drugs (methotrexate, phenytoin [Dilantin]). Risk factors for folate deficiency include poor diet (seen frequently in the poor, elderly persons, and people who do not eat fresh fruits or vegetables), alcoholism, history of malabsorption disorders, and pregnancy (especially the third trimester). Folate deficiency anemia occurs in about 4 of 100,000 people.
Pernicious anemia is caused by a deficiency of intrinsic factor, a substance secreted by the stomach parietal cells that is necessary for absorption of vitamin B12 (cobalamin). Because vitamin B12 may be stored for several years, this form of nutritional deficiency is rare and usually does not develop until late adulthood. Most often, it occurs in 40- to 70-year-old northern Europeans of fair complexion, with one notable exception. Early onset in black American women, 21% of whom were younger than 40 years of age, has been observed.
Long–standing pernicious anemia is associated with increased risk for development of gastric carcinoma. In addition, an association with myxedema, rheumatoid arthritis, and neuropsychiatric and neuromuscular abnormalities (due to a defect in myelin synthesis) has been reported.
Hemolytic Anemia
Hemolytic anemias are caused by immune attack, extrinsic factors (infection, splenomegaly, drugs, eclampsia), disorders of the RBC membrane (spherocytosis), enzymopathies (glucose-6-phosphate dehydrogenase [G-6-PD] deficiency), and hemoglobinopathies (sickle cell anemia, thalassemia). G-6-PD deficiency and sickle cell anemia are discussed here to illustrate the problems presented by the hemolytic anemias.
Hemolytic Anemia: Glucose-6-Phosphate Dehydrogenase Deficiency
The search during World War II for a substitute quinine led to the use of newer antimalarial drugs and the discovery of deficiency of glucose-6-phosphate dehydrogenase (G-6-PD), an enzyme that helps the RBC to turn carbohydrates into energy. This discovery occurred after several persons who were given primaquine developed hemolytic anemia because they lacked G-6-PD, an enzyme needed for the hexose monophosphate shunt pathway.
Glucose enters the RBC through a carrier mechanism, independent of insulin. About 90% of glucose is metabolized by the glycolytic pathway. The remaining glucose is metabolized by the hexose monophosphate shunt pathway. The byproduct of the glycolytic pathway is adenosine triphosphate, which provides energy for the cell. The byproduct of the hexose monophosphate shunt pathway is nicotinamide adenine dinucleotide phosphate (NADPH), which is used to reduce various cellular oxidants.
G-6-PD is the most common enzymopathy of humans.
Clinical features of G-6-PD deficiency involve acute intravascular hemolysis, which may be severe. Jaundice, palpitations, dyspnea, and dizziness may result. Infection is the event that most commonly triggers hemolysis in G-6-PD A deficiency. Drugs are the most common trigger for hemolysis in G-6-PD MED deficiency. Of more than 40 drugs that can induce hemolysis, those of significance in dental practice include acetylsalicylic acid, acetophenetidin (phenacetin), dapsone, ascorbic acid, and vitamin K. Fava bean ingestion is the most common dietary cause of hemolytic anemia in persons with G-6-PD deficiency.
Sickle Cell Anemia
Sickle cell hemoglobin (HbS) was the first Hb variant of the more than 600 inherited human Hb variants (hemoglobinopathies) to be recognized. Of these, more than 90% have single amino acid substitutions in the Hb chain. HbS is the result of substitution of a single amino acid—valine for glutamic acid—at the sixth residue of the β chain. In contrast, the thalassemias, another type of hemoglobinopathy, are caused by deletions or mutations of the α or β globin gene that result in a defect in globin synthesis (reduced or absent synthesis of one or more globin chains).
Sickle cell disorders are distinguished by the number of globin genes affected. The two most common types are sickle cell trait and sickle cell (disease) anemia. Sickle cell trait is the heterozygous state in which the affected person carries one gene for HbS. Approximately 8% to 10% of African Americans carry the trait. In western Africa, 25% to 30% of the population may be carriers. Sickle cell anemia is the homozygous state. A gene from each parent contributes to formation of the HbS molecule responsible for the disease. The RBC in sickle cell anemia becomes sickle-shaped when blood experiences lowered oxygen tension or decreased pH, or when the patient becomes dehydrated.
Distortion of the RBC into a sickled shape results from deoxygenation or decreased blood pH, causing partial crystallization of HbS, polymerization, and realignment of the defective Hb molecule (
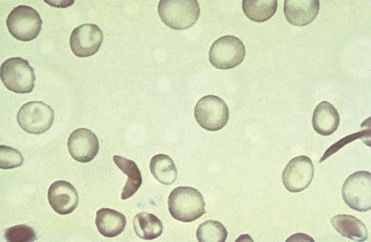
FIGURE 22-3 Sickle cell anemia. Peripheral blood smear shows characteristic abnormal sickle-shaped red blood cells (RBCs).
In patients with sickle cell anemia, more than 80% of the Hb is HbS. Clinical signs and symptoms of sickle cell anemia are the result of chronic anemia and small blood vessel occlusion. These manifestations include jaundice, pallor, dactylitis (hand and foot warmth and tenderness), leg ulcers, organomegaly, cardiac failure, stroke, attacks of abdominal and bone pain (aseptic necrosis), and delays in growth and development (
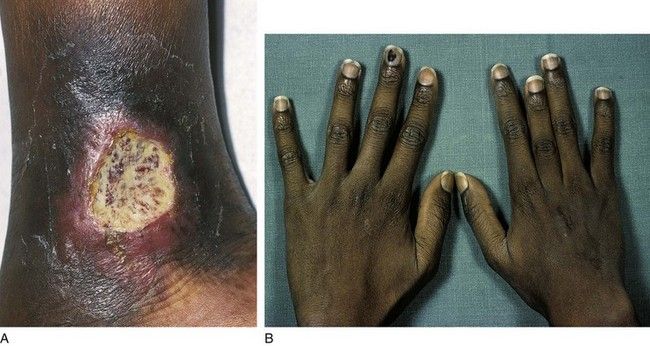
FIGURE 22-4 Sickle cell anemia may cause various complications. A, Leg ulcer secondary to a vasoocclusive attack. B, Growth deformation of the middle finger from dactylitis of the growth plate.
(From Hoffbrand AV, Pettit JE: Color atlas of clinical hematology, ed 4, London, 2010, Mosby.)
Complications of sickle cell anemia can occur at any age, but patients in the following age groups are more likely to manifest certain complications:
1 Birth to 20 years of age: painful events, stroke, acute chest syndrome (fever, chest pain, wheezing, cough, and hypoxia), acute anemia and infection
2 From 20 to 40 years of age: osteonecrosis of hip and shoulder joints, leg ulcers, priapism, liver disease, and gallstones
3 Older than 40 years of age: pulmonary hypertension, nephropathy, proliferative retinopathy, and cardiac enlargement, heart murmurs, and sudden death due to arrhythmias
People with the sickle cell trait generally have no symptoms unless they encounter situations in which abnormally low concentrations of oxygen are present (e.g., in an unpressurized airplane, through the injudicious administration of general anesthesia). Patients with sickle cell trait are much more resistant to sickling stimuli, because only 20% to 45% of their Hb is HbS. Patients with sickle cell trait are not at risk for adverse events during dental treatment unless severe hypoxia, severe infection, or dehydration also is present.
Aplastic Anemia
Aplastic anemia occurs when the bone marrow is unable to produce adequate numbers of RBCs, white blood cells, and platelets. The hematopoietic stem cells are unable to proliferate, differentiate, or give rise to mature blood cells.
Some cases of aplastic anemia are caused by drugs, viruses, organic compounds, and radiation. A few, such as Fanconi’s anemia, are inherited. A majority of cases, however, are idiopathic (50% to 65%).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


