Syndromes of the Head and Neck
A multitude of anomalies and syndromes occur in the head and neck, most of which are beyond the scope of this book. The chapters in this section cover some of the most common anomalies or syndromes associated with the craniomaxillofacial region (Table 14-1). Congenital anomalies include nonsyndromic craniosynostosis, hemifacial microsomia, and cleft lip and palate; congenital syndromes include Apert and Crouzon. Obstructive sleep apnea syndrome, although not congenital in nature, is included in this section because its pathogenesis and manifestation are based on anatomic anomalies of the head and neck. Oral and maxillofacial surgeons are uniquely trained to play an integral role in the surgical management of these patients, syndromic or not.
Cleft Lip and Palate
Examination
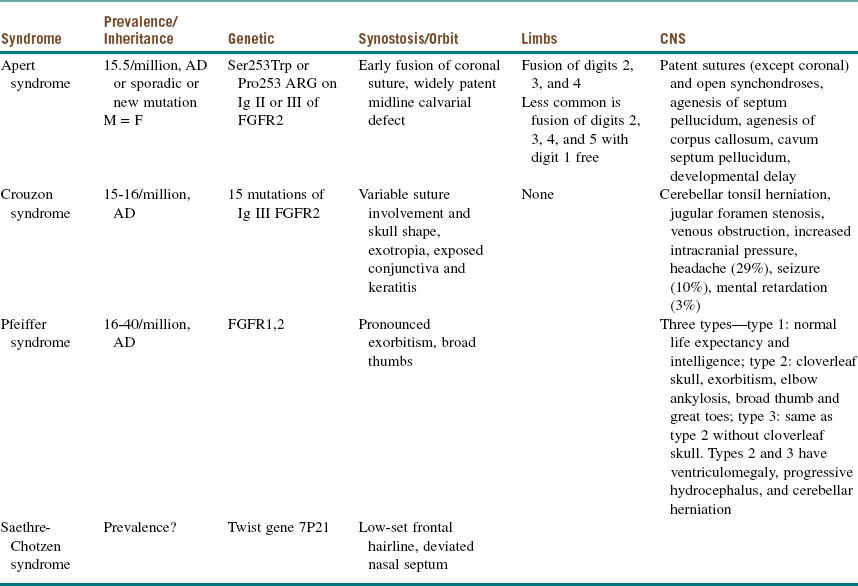
Maxillofacial. The cleft lip (CL) is complete, penetrating the entire thickness of the lip, alveolus, nasal tip cartilages, and floor of the nose (Figure 14-1). The cleft is unilateral, right of midline (left side prevalence), and continuous with the palate (CLP is most commonly expressed unilaterally, with a 2 : 1 predilection for the left side).
Treatment
Table 14-2 outlines the sequence of management of patients with CLP. CL repair is usually addressed at 10 to 14 weeks of age. One advantage of waiting until this age is that it allows time for a thorough medical evaluation to determine whether the infant has any congenital defects. The surgical procedure is generally easier to perform when the child is slightly larger, because anatomic landmarks are more prominent and well defined. In addition, it has historically been accepted that the safest anesthesia time period for infants is based on the “rule of tens”—surgery can be performed when the child is at least 10 weeks of age, weighs at least 10 pounds, and has a minimum hemoglobin value of 10 mg/dl (however, there is no current scientific rationale to support this rule). With modern intraoperative pediatric monitoring techniques, general anesthesia can be performed safely at an earlier age as needed, although there is no documented benefit to performing lip repair before 3 months of age. In addition, excessive scarring and inferior esthetic results have been found to occur when surgery is performed earlier than age 3 months.
Table 14-2
Sequence of Management of the Cleft Palate
Modified from Kaban LB, Troulis MJ (eds): Pediatric oral and maxillofacial surgery, St Louis, 2004, Saunders.
Many techniques have been devised for CP repair. The Bardach technique involves creation of two large, full-thickness flaps on each palatal shelf, which are layered and brought to the midline for closure (Figure 14-2, A and B). This technique allows for preservation of the palatal neurovascular bundle, which is contained within the pedicle of each flap. The Von Langenbeck technique is similar to the Bardach technique, but it preserves an anterior pedicle for increased blood supply to the flaps. It also involves elevation of large mucoperiosteal flaps from the palate with midline approximation of the cleft margins. Long lateral releasing incisions are made at the border of the palatal and alveolar bone to allow mobilization. The levator muscles are detached from their abnormal insertion along the hard palate. The Furlow double-opposing Z-plasty technique involves two Z-plasties, one on the oral mucosa and one in the reverse orientation on the nasal mucosa (Figure 14-2, C). The levator muscle on one side is included in the posteriorly based oral mucosa Z-plasty, whereas the levator muscle from the opposite side is included within the posteriorly based nasal mucosal Z-plasty flap. This procedure produces palatal lengthening and reorients and provides overlap of the malpositioned levator muscles. The Furlow Z-plasty has been reported to be associated with a higher rate of fistula formation at the junction of the soft and hard palates.
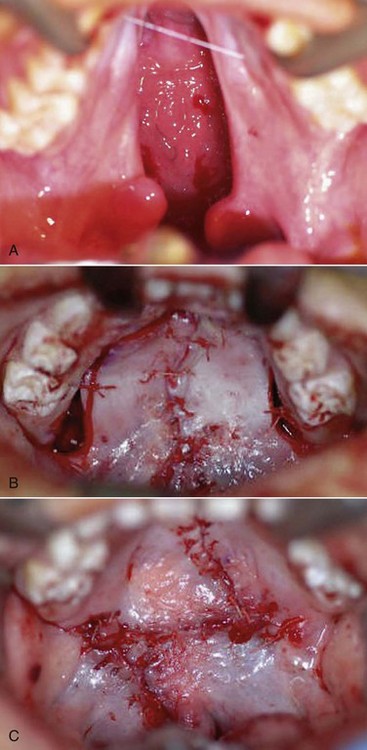
Figure 14-2 A, Complete cleft palate. B, Immediate postoperative photograph showing cleft palate closure using the Bardach technique. C, Immediate postoperative photograph showing cleft palate closure using the Furlow double-opposing Z-plasty technique.
Figure 14-3 shows the 1-year postoperative view of the patient seen in Figure 14-1.
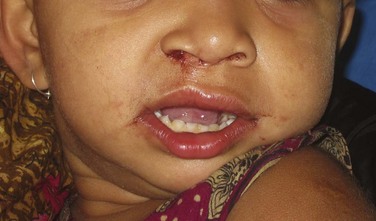
Figure 14-3 Postoperative photograph of patient in Figure 14-1 after lip and palate repair at 1 year of age. (Courtesy of Dr. Shahid Aziz.)
Discussion
A number of concerns are associated with CLP.
• Feeding. Approximately 25% of CLP infants have early feeding difficulties, with poor weight gain for the first 2 to 3 months. Feeding sessions are prolonged due in part to ulceration of the nasal mucosa. Some infants also have increased metabolic needs due to congenital heart disease or airway obstruction. Initial poor weight gain usually resolves after cleft closure, and any deficiency in growth is corrected by 6 months of age. Height and weight progressions are routinely monitored.
• Speech and language development. Even after the palate has been repaired, children are still at risk for subsequent speech disorders. It is reported that 25% of children with CLP develop normal speech after primary surgery, whereas the remaining 75% require many surgical interventions throughout childhood and adolescence. Speech problems arise from velopalatal insufficiency, dental and occlusal problems, oronasal fistulas, and hearing problems. Approximately 15% to 20% of patients who have CP repair within the first 12 to 15 months of life have velopharyngeal insufficiency. As mentioned earlier, surgical intervention must be coordinated with the development of speech. Speech and language therapy must also be provided during this time. The monitoring of speech continues into adolescence and adulthood in conjunction with active orthodontic and surgical management.
• Hearing. Patients with CP are at increased risk of middle ear effusions and subsequent infections. The attachment of the levator veli palatini muscle around the eustachian tube is abnormal and leads to poor aeration and drainage of the middle ear. Regular assessment by the ear, nose, and throat (ENT) surgeon and audiologist is recommended to ensure that poor hearing is not a contributing factor to compromised speech.
• General dental welfare. Children with CLP are at great risk for developing malocclusion. When the cleft involves the alveolar process, odontogenic structures within this region are routinely absent or malformed. Orthodontic intervention is generally initiated during the preschool years. Active occlusal manipulation and correction should not be instituted until the permanent dentition has erupted.
• Genetics. There are three types of genetic risk groups for CLP: the syndromic group, identified by physical examination; the familial group, identified by history; and isolated defects, identified by exclusion of the first two groups. As mentioned earlier, the incidence of CL is approximately 1 : 700 live births, and the incidence of CP is approximately 1 : 2,000 live births. With one parent and one child affected, the chance of a second child having a cleft is 10%. When both parents are without clefts and two children have clefts, the chance of a third child having a cleft is 19%. When one parent has a cleft and two offspring are normal, the chance of the third child being born with a cleft is 2.5%.
• Environment. Epidemiologic studies have demonstrated a relationship between maternal exposure to environmental factors or teratogens during pregnancy and the development of CLP. These factors or teratogens include alcohol consumption, cigarette smoking, folic acid deficiency, corticosteroids, benzodiazepines, and anticonvulsants.
Bryne, PJ, Sands, NR. Secondary bone grafting of residual alveolar and palatal clefts. J Oral Surg. 1972; 30:87–92.
Carici, F. Recent developments in orofacial cleft genetics. J Craniofac Surg. 2003; 142(2):130–143.
Chung, K, Kowalski, C, Kim, HM, et al. Maternal cigarette smoking during pregnancy and the risk of having a child with cleft lip/palate. Plast Reconstr Surg. 2000; 105(2):485–491.
Copeland, M. The effect of very early palatal repair on speech. Br J Surg. 1990; 43:676.
Costello, BJ, Ruiz, RL, Turvey, T. Surgical management of velopharyngeal insufficiency in the cleft patient. Oral Maxillofac Surg Clin North Am. 2002; 14:539–551.
Costello, BJ, Shand, J, Ruiz, RL. Craniofacial and orthognathic surgery in the growing patient. Select Readings Oral Maxillofac Surg. 2003; 11(5):1–20.
Dorf, DS, Curtin, JW. Early cleft repair and speech outcome: a ten year experience. In: Bardach J, Morris HL, eds. Multidisciplinary management of cleft lip and palate. Philadelphia: Saunders; 1990:341–348.
Glenny, AM, Hooper, L, Shaw, WC, et al. Feeding interventions for growth and development in infants with cleft lip, cleft palate or cleft lip and palate. Cochrane Database Syst Rev. 2, 2005.
Habel, A, Sell, D, Mars, M. Management of cleft lip and palate. Arch Dis Child. 1996; 74(4):360–366.
Lees, VC, Pigott, RW. Early postoperative complications in primary cleft lip and palate surgery: how soon may we discharge patients from hospital? Br J Plast Surg. 1992; 45:232–234.
Marsh, JL. Craniofacial surgery: the experiment on the experiment of nature. Cleft Palate Craniofac J. 1996; 33:1.
McComb, H. Primary correction of unilateral cleft lip nasal deformity: a ten year review. Plast Reconstr Surg. 1985; 75:791–799.
Murray, JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002; 61(4):248–256.
Padwa, BL, Sonis, A, Bagheri, SC, et al. Children with repaired bilateral cleft lip/palate: effect of age at premaxillary osteotomy on facial growth. Plast Reconstr Surg. 1999; 105:1261.
Posnick, JC. Cleft orthognathic surgery: the unilateral cleft lip and palate deformity. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: Saunders; 2000:860–907.
Posnick, JC. The staging of cleft lip and palate reconstruction: infancy adolescence. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: Saunders; 2000:785–826.
Ruiz, RL, Costello, BJ, Turvey, T. Surgical correction of midface deficiency in cleft lip and palate malformation. Oral Maxillofac Surg Clin North Am. 2002; 14(4):491–507.
Spritz, RA. The genetics and epigenetics of orofacial clefts. Curr Opin Pediatr. 2001; 13(6):556–600.
Takato, T, Yonohara, Y, Mori, Y. Early correction of the nose in unilateral cleft lip patients using an open method: a ten year review. J Oral Maxillofac Surg. 1995; 53A:28–33.
Nonsyndromic Craniosynostosis
HPI
The mother of this 7-month-old, otherwise healthy male (craniosynostosis has a male predilection) has been concerned about the abnormal shape of his head, which was noticed immediately after birth. (Craniosynostosis is the premature fusion of the cranial sutures during intrauterine life. The deformity is often noticeable early [Figure 14-4].) The pediatrician has been closely observing this skull deformity for changes and resolution. It was initially assumed to be deformational plagiocephaly (skull deformity caused by vaginal delivery or early fetal decent into the pelvis) and later was thought to be secondary to a positional plagiocephaly, an acquired skull deformity caused by a repetitive head position during sleep (nonsynostotic posterior plagiocephaly has increased since the American Academy of Pediatrics issued a recommendation that infants be placed on their back during sleep to reduce the risk of sudden infant death syndrome [SIDS]). Despite conservative management, the child continued to exhibit the cranial deformity, which appeared to slightly worsen over time. He is otherwise in good health, and the mother denies any behavioral abnormalities. He is referred for craniofacial evaluation (the rate of detectable cranial abnormalities secondary to craniosynostosis has been reported to be as high as 1 : 1,700 to 1 : 1,900 births).
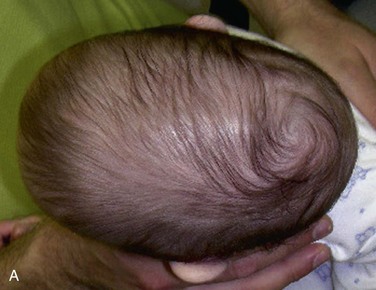
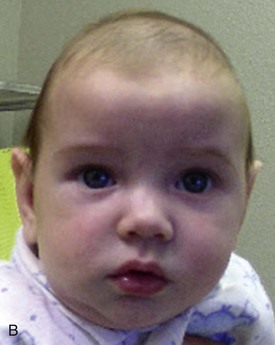
Examination
General. The patient is a well-developed and well-nourished, pleasant child in no apparent distress.
Complications
Despite the very low complication rates associated with craniofacial surgery, both intraoperative and postoperative complications can occur. Massive blood loss and postoperative infection are the most common and most feared complications (Box 14-1). There is a high likelihood that homologous blood transfusion will be required during cranial vault reshaping. This is in part due to the low effective blood volume in infants and children. Acute normovolemic hemodilution (ANH) is a technique commonly performed intraoperatively to reduce the need for transfusion. Additional techniques include intraoperative blood salvage. Maintaining a “safe” hematocrit level, between 28% and 35%, has been recommended. However, the incidence of transfusion has been estimated to be as high as 90%.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


