13
Haematology
13A General haematology, haemato-oncology
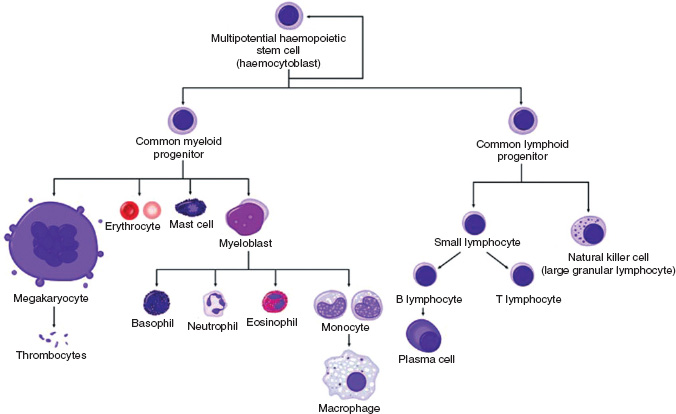
Normal haemopoiesis
Haemopoiesis is the term referring to the formation of blood cells. In humans it occurs in different sites during foetal development and following birth. During the first 8 weeks of gestation, the yolk sac is the main site of haemopoiesis. Between 2 and 7 months of gestation, the liver and spleen take over blood production, and from 7 months gestation the bone marrow is the major site. The bone marrow remains the chief source of haemopoiesis throughout adulthood, with haemopoietic tissue found within the central skeleton and proximal long bones of adults.
All blood cells originate from a common pluripotential stem cell that is capable of self-renewal. Stem cells undergo differentiation to produce the early progenitor cells of each of the cell lineages. As further differentiation and maturation occur, cells become haemopoietic progenitors committed to a single cell lineage, ultimately resulting in the formation of granulocytes, erythrocytes, monocytes and macrophages, megakaryocytes, eosinophils, basophils, and T and B lymphocytes (Fig. 13A.1).
The bone marrow stroma
Bone marrow stromal cells form a specialised microenvironment for stem cell growth and differentiation. Adipocytes, fibroblasts, macrophages and other cells secrete substances such as collagen, glycoproteins and glycosaminoglycans which form an extracellular matrix on which stem cells can grow. The stromal cells also secrete haemopoietic growth factors which stimulate differentiation and maturation of blood cells. Adhesion molecules produced by the stromal cells fix the marrow precursor cells to the extracellular matrix, enabling them to mature under the influence of the growth factors.
Erythropoiesis
The stem cell differentiates to form the early mixed myeloid progenitor cell, followed by BFU-E (burst-forming unit erythroid) and then CFU-E (colony-forming unit erythrocyte) from which the pronormoblast arises. This large cell has a basophilic (blue) cytoplasm and a large central nucleus with clumped chromatin. Successive smaller normoblasts develop within the marrow. These contain increasing amounts of haemoglobin in the cytoplasm and therefore stain increasingly pink. Finally, in the marrow, the nucleus is extruded from the late normoblast to form a reticulocyte. The reticulocyte contains ribosomal ribonucleic acid (rRNA) and is released into the peripheral blood for 1–2 days before losing its RNA to form a mature erythrocyte. Red cells are nonnucleated biconcave discs which survive for 120 days in the peripheral blood before they are broken down by macrophages belonging to the reticuloendothelial system.
The haemopoietic growth factor that controls erythropoiesis is erythropoietin, a hormone produced by the kidney. In anaemia, oxygen levels in renal tissue fall, stimulating erythropoietin production. This in turn increases erythropoiesis within the bone marrow. Conversely, in states of increased red cell mass (polycythaemia), the increased oxygen tension in the kidneys reduces erythropoietin production and reduces erythropoiesis.
Figure 13A.1 Normal haemopoiesis.
Haemoglobin
The primary role of red blood cells is to transport oxygen to the body tissues via the systemic arterial system and to carry carbon dioxide from the tissues to the lungs in the venous system. Red cells transport oxygen and carbon dioxide via the specialised protein they contain called haemoglobin (Hb).
A normal Hb molecule is composed of two pairs of polypeptide chains, each of which encloses an iron-containing porphyrin called haem, which can bind reversibly with oxygen. The relationship between the four globin chains is known as the quaternary structure and can change depending on whether or not the haemoglobin molecule is oxygenated. When oxygen is released from Hb, the globin chains slide on each other, allowing 2,3-diphosphoglycerate (2,3-DPG) to enter the Hb molecule, reducing its affinity for oxygen. Oxygenation of Hb pulls the β-chains together and 2,3-DPG is extruded from the Hb molecule.
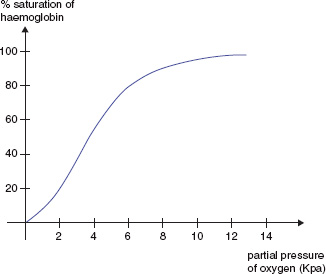
Changes in the quaternary structure are responsible for the sigmoid shape of the Hb oxygen dissociation curve (Fig. 13A.2). Hypoxia increases synthesis of 2,3-DPG pushing the oxygen dissociation curve to the right, resulting in increased offloading of oxygen to the tissues. Acidosis, pyrexia and high carbon dioxide concentrations also push the curve to the right (the Bohr effect).
Figure 13A.2 The haemoglobin oxygen dissociation curve.
The predominant Hb produced during foetal life is ‘foetal haemoglobin’ (HbF) which contains two α-globin chains and two γ-globin chains. At 3–6 months of age, infants switch to producing ‘adult haemoglobin’ (HbA; two α- and two β-chains).
Although HbA is the predominant Hb after 6 months of age, small quantities of HbF and HbA2 (two α- and two δ-chains) persist into adulthood.
Red cell metabolism
The ‘Embden–Meyerhof’ pathway is an anaerobic glycolytic pathway within the red cell that generates energy in the form of adenosine triphosphate (ATP). ATP helps to maintain red cell volume, shape and flexibility. Reduced nicotinamide adenine dinucleotide (NADH) is also formed during glycolysis and is needed to convert functionally inactive metHb containing oxidised iron (Fe3+) to functionally active Hb which contains ferrous iron (Fe2+).
Red cells generate reduced nicotinamide adenine dinucleotide phosphate (NADPH) via the hexose monophosphate pathway. NADPH is used to generate reduced glutathione which protects the cell from oxidant stress. 2,3-DPG is also produced by the red cell to regulate the oxygen affinity of haemoglobin.
Anaemia
Anaemia is a reduction in the Hb concentration of the blood. Although normal reference ranges for Hb concentration vary between laboratories, in general a Hb <2 11.5 g/dl in a female and <13 g/dl in a male is considered low.
Anaemias may be classified according to red cell indices (Table 13A.1). The mean corpuscular volume (MCV) is the volume of the average red blood cell. Mean corpuscular haemoglobin (MCH) is the Hb content of the average red cell.
General features of anaemia
A fall in Hb concentration leads to a reduction in the oxygen-carrying capacity of the blood, resulting in tissue hypoxia. The body attempts to compensate by increasing stroke volume and heart rate, thereby increasing cardiac output. Young, fit patients may experience few symptoms and signs apart from pallor (due to vasoconstriction of blood vessels in the skin) fatigue, tachycardia and, occasionally, development of a heart murmur due to a hyperdynamic circulation. Older patients may not tolerate the anaemia, however, and can develop additional features such as palpitations, dyspnoea, ischaemic symptoms such as angina and signs of cardiac failure.
Table 13A.1 Classification of anaemias according to red cell indices
| Hypochromic microcytic | Normochromic normocytic | Macrocytic |
| Iron deficiency Thalassaemia Lead poisoning Hereditary sideroblastic anaemia Anaemia of chronic disease (some cases) |
Anaemia of chronic disease (some cases) Renal failure Acute blood loss Haemolysis Bone marrow infiltration Mixed deficiencies |
Megaloblastic anaemias Liver disease Alcohol excess Pregnancy Hypothyroidism Myelodysplasia Aplastic anaemia Multiple myeloma Reticulocytosis |
Iron deficiency anaemia
Iron absorption and distribution
A normal diet contains approximately 10 mg of elemental iron but usually only 10% of the iron is absorbed. Dietary iron is converted from the ferric form (Fe3+) to the ferrous form (Fe2+) by acid in the stomach. Fe2+ binds to transferrin which is produced by duodenal epithelial cells. Bound iron is absorbed mainly in the duodenum, but also in the jejunum. Absorbed iron is used to form the haem part of haemoglobin and is also found in myoglobin in muscles and in liver enzymes. The remainder is stored in the liver, bone marrow and spleen as ferritin and haemosiderin.
Causes of iron deficiency
The most common cause of iron deficiency is chronic blood loss, which may occur from any site. A common site is the gastrointestinal (GI) tract where inflammation, ulceration or malignancy may be the source of blood loss. Menorrhagia and haematuria may also cause iron deficiency. Other causes are listed in Table 13A.2.
Clinical features of iron deficiency anaemia
In addition to the general features of anaemia, patients with iron deficiency may also develop specific signs such as koilonychia (concave nails), generalised pruritus, pica (cravings for unusual foods), angular stomatitis and painless glossitis. In children, psychomotor delay and cognitive decline can occur. Rarely, adults may present with dysphagia and iron deficiency due to the presence of a pharyngeal web, which is associated with oesophageal cancer (Plummer–Vinson or Paterson–Kelly syndrome).
Table 13A.2 Causes of iron deficiency
|
|
|
|
Laboratory investigation of suspected iron deficiency
- In the full blood count (FBC) a low Hb, MCV and MCH are seen. The platelet count may be moderately raised, especially if there is ongoing blood loss
- In the blood film, hypochromic, microcytic red cells are seen. Anisocytosis (variation in red cell size), pencil-shaped red cells and target cells are also present
- Iron studies shows low serum iron, raised total ironbinding capacity (TIBC), low serum ferritin, transferrin saturation (serum iron/TIBC) <10% and raised serum transferrin receptor level
- Antiendomysial antibodies are assayed to screen for coeliac disease.
Treatment
Iron replacement (oral or parenteral) is required and the underlying cause should be investigated.
Megaloblastic anaemias
Megaloblastic anaemia results from deficiency of vitamin B12 or folic acid. Vitamin B12 and folate are needed for synthesis of deoxyribonucleic acid (DNA) within the nucleus of maturing erythroblasts. Deficiency of either causes delayed maturation of the nucleus compared with the cytoplasm, resulting in the formation of megaloblasts in the marrow.
Absorption of vitamin B12
Vitamin B12 is found in foods of animal origin and binds to intrinsic factor, which is produced by gastric parietal cells. The intrinsic factor–B12 complex is absorbed in the terminal ileum. B12 binds to transcobalamin I in the blood but it is transcobalamin II that is responsible for delivery of B12 to the peripheral tissues. Table 13A.3 highlights the common causes of vitamin B12 deficiency.
Table 13A.3 Causes of vitamin B12 deficiency
|
|
|
Table 13A.4 Causes of folate deficiency
|
|
|
|
Folate deficiency
Folic acid is found in leafy vegetables and is absorbed mainly in the duodenum. The common causes of folate deficiency are listed in Table 13A.4.
Clinical features of megaloblastic anaemia
Both B12 and folate deficiency can cause angular stomatitis, painful glossitis and mild jaundice. In severe cases infertility may occur. Specifically, vitamin B12 deficiency can cause demyelination of the dorsal columns and corticospinal tracts in the spinal cord, along with peripheral nerve damage. This is known as ‘subacute combined degeneration of the cord’. Patients may present with ataxia due to loss of proprioception and vibration sense. Corticospinal tract lesions in combination with a peripheral neuropathy classically cause extensor plantar responses, brisk knee reflexes and absent ankle reflexes. Rarely, cognitive decline and visual loss also occur.
Laboratory investigations
- FBC shows low Hb, high MCV
- Blood film shows typically oval macrocytes, and hypersegmented neutrophils are seen
- Measurement of serum B12 and red cell folate shows one or both to be low
- Intrinsic factor antibodies should be checked if vitamin B12 deficiency is found
- Bone marrow aspirate (not usually required for diagnosis) demonstrates a hypercellular marrow. Megaloblasts and giant metamyelocytes are seen in the marrow
- Schilling test – radiolabelled B12 is ingested and, if absorbed, is excreted in the urine. Unlabelled B12 is given as an intramuscular injection to saturate tissue stores. In malabsorptive states, <5% of the radiolabelled B12 will be detected in the urine. The test is then repeated with the addition of intrinsic factor. If the addition of intrinsic factor results in normal B12 levels in the urine, the malabsorption must be due to pernicious anaemia rather than an intestinal cause.
Treatment
Replacement with folic acid and/or vitamin B12 is required. If folate deficiency is detected, B12 deficiency must always be excluded and, if present, corrected before folate replacement is given, as folate replacement alone may precipitate subacute combined degeneration of the cord. The reticulocyte count, which begins to rise 2–3 days after commencing treatment and peaks at 6–7 days, may be used to monitor response to treatment. B12 replacement in patients with dorsal column involvement can partly improve the peripheral neuropathy but the spinal cord damage is irreversible.
Anaemia of chronic disease
A normochromic, normocytic anaemia (occasionally hypochromic, microcytic) can be seen in patients with chronic inflammatory and malignant conditions. Serum iron is low despite adequate iron stores in the bone marrow. TIBC is also low, and serum ferritin may be normal or raised as ferritin is an acute phase protein that rises in response to inflammation. Increases in cytokines such as tumour necrosis factor and interleukin-1 lead to a blunted marrow response to erythropoietin, impaired iron uptake into erythroid precursors and reduced red cell survival. In addition, hepcidin released from the liver in response to inflammation inhibits the release of iron from macrophages into the plasma. Treatment of the underlying condition may improve the anaemia. Patients with renal failure and some malignant conditions may also benefit from treatment with recombinant erythropoietin.
Haemolytic anaemias
Haemolysis is the increased destruction of red cells. Red cell destruction may be:
- Intravascular – lysis of red cells within blood vessels due to complement activation
- Extravascular – antibody binds to red cells. Antibodycoated red cells are then removed from the circulation by macrophages within the liver and/ or the spleen.
If haemolysis is mild, the marrow is able to compensate by increasing red cell production. Erythroid hyperplasia in the marrow causes a reticulocytosis. Reticulocytes are visible as polychromatic erythrocytes (larger than normal red cells which stain slightly blue) on the blood film. In severe haemolysis, the marrow cannot compensate fully and the patient becomes anaemic.
In addition to the general features of anaemia, patients with haemolysis become jaundiced due to the release of unconjugated bilirubin from lysed red cells. Extravascular haemolysis often causes splenomegaly.
All patients with haemolytic anaemia are at risk of developing acute marrow aplasia if they become folate deficient or develop parvovirus B19 infection. Marrow production of red cells is switched off, causing profound anaemia with a reticulocytopaenia, often requiring red cell transfusion.
Classification of haemolytic anaemias
Haemolytic anaemias may be hereditary or acquired (Table 13A.5).
Laboratory investigation of haemolysis
- FBC shows low Hb; the MCV is often raised due to the presence of large numbers of reticulocytes in the blood
- In the blood film, red cell fragments are seen in intravascular haemolysis. Spherocytes (small, densely staining red cells with no central pallor) are seen in extravascular haemolysis
- A raised lactate dehydrogenase (LDH) reflects high cell turnover
- There is raised unconjugated bilirubin
- There are low serum haptoglobins – haptoglobins are plasma proteins which bind the polypeptide chains released from the breakdown of Hb. Bound haptoglobin is then taken up by cells of the reticuloendothelial system
- The direct antiglobulin test (DAT) detects antibody bound to the surface of red cells. A positive DAT is seen in autoimmune haemolytic anaemia.
Table 13A.5 Causes of haemolytic anaemia
| Hereditary | Acquired |
| Red cell membrane abnormalities, e.g. hereditary spherocytosis, hereditary elliptocytosis Defects in red cell metabolism, e.g. glucose-6-phosphate dehydrogenase deficiency Haemoglobinopathies, e.g. sickle cell anaemia |
Autoimmune Alloimmune, e.g. haemolytic transfusion reaction Red cell fragmentation syndromes, e.g. thrombotic thrombocytopaenic purpura (TTP) Renal and liver disease Burns Drugs causing oxidative haemolysis, e.g. dapsone Paroxysmal nocturnal haemoglobinuria Infection, e.g. malaria |
Red cell membrane defects
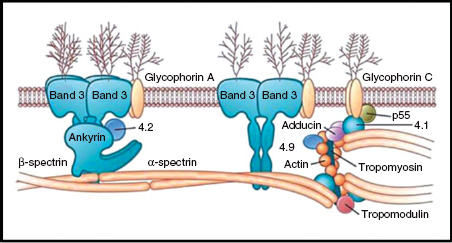
The red cell membrane is made up of a phospholipid bilayer, integral proteins and a cytoskeleton located on the inner aspect of the lipid bilayer (Fig. 13A.3). The cytoskeleton is composed of proteins such as α- and β-spectrin, actin and protein 4.1 which maintain red cell shape and flexibility. These structural proteins connect to the transmembrane protein band 3 via ankyrin and protein 4.2. The most common hereditary haemolytic anaemia in Northern Europe is hereditary spherocytosis (HS) and it is caused by defects in these ‘vertical connections’.
Hereditary spherocytosis (HS)
HS is usually inherited as an autosomal dominant condition. The majority of patients with HS have mutations of spectrin and ankyrin genes resulting in defects in the ankyrin–spectrin complex. About 25% of patients have a deficiency of band 3 protein. A smaller number have a deficiency of protein 4.2 or the defect is unknown. Disruption of these ‘vertical connections’ in the red cell membrane results in the formation of spherocytes, which are destroyed early by the spleen. The clinical severity of the condition varies from mild to severe, and patients with moderate or severe haemolysis usually require splenectomy.
Autoimmune haemolytic anaemia
Autoimmune haemolytic anaemia (AIHA) is an acquired form of haemolysis in which autoantibodies directed against red cell antigens attach to red cells, resulting in increased erythrocyte destruction. If the autoantibody attaches at body temperature it is classed as a warmacting antibody. These antibodies are usually polyclonal IgG antibodies and cause a strongly positive DAT.
First-line treatment of warm AIHA is high dose oral prednisolone. Red cell transfusion is only given if the Hb is dangerously low or the patient’s cardiovascular system is compromised by the anaemia.
Cold-acting autoantibodies are usually IgM antibodies that bind to erythroctyes in the peripheral circulation at lower temperatures and may cause red cell agglutination. Cold haemagglutinin disease (CHAD) is the most common form of cold AIHA. The idiopathic form of CHAD is more common in older patients and often runs a chronic course. CHAD may also occur following an infection when polyclonal IgM forms as a response to the infection but cross-reacts with red cell antigens. Mild haemolysis usually develops 2–3 weeks after the infection and is self-limiting. Rarely, severe intravascular haemolysis occurs following Mycoplasma pneumoniae infection. CHAD may also be associated with lymphoproliferative disorders.
Haemoglobinopathies
Mutation of globin genes or the genes that control their expression can result in changes in the amino acid sequence of globin, resulting in the synthesis of structurally abnormal or ‘variant’ Hb. A variant Hb may possess altered oxygen affinity, solubility or stability compared with normal Hb. Globin gene mutation can also result in a reduced rate of synthesis of globin chains (thalassaemia). Mutation of the α- and β-globin genes causes α- and β-thalassaemia, respectively. The clinical severity of the condition corresponds to the number of genes affected.
Figure 13A.3 Structure of the red cell membrane.
Sickle cell disease
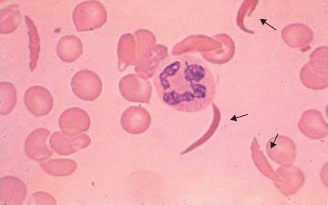
Sickle cell haemoglobin (HbS) is an example of a β-chain variant in which mutation of the β-globin gene results in substitution of valine for glutamic acid at position 6 of the β-globin chain. On deoxygenation, the solubility of HbS is reduced and polymerisation occurs. This distorts the red cell into a ‘sickle’ shape (Fig. 13A.4). The deformed red cells occlude capillaries, leading to tissue hypoxia.
The sickle mutation is prevalent in Africa, the Mediterranean, the Americas and Asia. Homozygosity for haemoglobin S (HbSS) results in sickle cell disease. Heterozygosity (HbAS) is known as ‘sickle cell trait’ and is asymptomatic, but the mutated β-gene may be inherited by children. Sickle cell trait affords some protection against infection with Plasmodium falciparum. The mutated β-globin gene may also be co-inherited with other β-chain variants, causing clinical features of sickle cell disease (SCD).
Clinical features of sickle cell disease
Dehydration, hypoxia or exposure to the cold or infection precipitates painful sickling of red cells, leading to vascular occlusion. Renal infarction can occur with eventual progression to renal failure. Retinal ischaemia can jeopardise vision. Avascular necrosis or osteomyelitis can result from bony infarcts, and infarction of the skin can lead to ulceration. Recurrent pulmonary infarcts may lead to the development of pulmonary hypertension. In children, pooling of red cells in a rapidly enlarging spleen causes acute anaemia and is known as ‘splenic sequestration’. A life-threatening complication of SCD is the ‘acute chest syndrome’. It is characterised by acute hypoxia, fever, chest pain and pulmonary infiltrates on the chest radiograph. The hypoxia can cause widespread sickling, leading to organ failure. Cerebral haemorrhage and infarction are also significant causes of morbidity. All patients are at increased risk of infection as they become functionally hyposplenic following recurrent splenic infarction. In addition, chronic haemolysis leads to the formation of pigment gallstones, and symptomatic patients should undergo cholecystectomy.
Figure 13A.4 Sickle cells.
Acute management of a painful crisis
Painful sickling requires rehydration with intravenous fluids, oxygen support, opiate analgesia and antibiotics if infection is suspected. Prophylactic dose low molecular weight heparin (LMWH) is given to prevent thrombotic events. Red cell transfusion or exchange transfusion may be required.
α-Thalassaemia
Reduced rate of production of α-globin is usually caused by deletions of one or more of the four α-genes on chromosome 16. Occasionally α-thalassaemia may be non-deletional, due to point mutations causing gene dysfunction or due to mutation in genes controlling α-gene expression.
Pathophysiology
Reduced production of α-globin results in an excess of γ- or β-chains that form tetramers (HbH and Hb Barts). HbH is unstable and precipitates out in red cells, forming red cell inclusions. Chronic extravascular haemolysis ensues as red cells are removed from the circulation by the spleen. In addition, HbH has a very high affinity for oxygen, resulting in reduced offloading of oxygen to the tissues.
‘α-Thalassaemia trait’ is caused by deletion of either one or two of the α-genes. Patients often have a normal blood count or a mild hypochromic, microcytic anaemia and raised red cell count. Loss of three genes causes HbH disease, characterised by a moderate hypochromic, microcytic anaemia and splenomegaly. HbH comprises tetramers of β-chains. Loss of all four genes leads to total failure of α-chain synthesis so that no synthesis of HbF, A or A2 can occur. Hb Barts forms in the foetus and is composed of four γ-chains. A severe anaemia develops, causing cardiac failure and a hydropic foetus. Death usually occurs in utero or shortly after birth.
β-Thalassaemia
Pathophysiology
Reduced or absent β-chain production results in an excess of α-chains, which complex with γ- or δ-chains to form HbF and HbA2, respectively. The remaining α-chains precipitate out in developing erythroblasts, causing them to undergo apoptosis within the marrow, resulting in ‘ineffective erythropoiesis’. Some mature red cells do enter the peripheral blood but, as they contain α-chain inclusions, they are removed by the spleen. The combination of ineffective erythropoiesis and haemolysis leads to anaemia. Anaemia, along with the high level of HbF, stimulates erythropoietin production and the marrow expands to compensate.
There are two allelic β-globin genes on chromosome 11. β-Thalassaemia results from mutation of one or both β-genes or mutation of a gene controlling β-gene expression. β-Thalassaemia heterozygosity (‘β-thassaemia trait’) is symptomless and carriers have a mild hypochromic, microcytic anaemia, raised red cell count and raised HbA2.
Homozygosity or compound heterozygosity for β-thalassaemia (‘β-thalassaemia major’) produces a clinically severe phenotype. At 3–6 months of age, the switch from γ- to β-chain synthesis cannot occur and anaemia develops. If untreated, marrow expansion causes bony deformity, such as bossing of the skull. Chronic haemolysis causes hepatosplenomegaly. These features can be largely avoided by regular red cell transfusion. Repeated transfusion leads to iron overload, however, and iron deposition in the liver, heart and endocrine organs. Without long-term iron chelation therapy, patients die young of cardiac failure or cardiac arrhythmias.
Red cell fragmentation syndromes
Fragmentation of red cells can occur when they come into contact with abnormal surfaces or turbulent blood flow. Common causes of physical damage to red cells are prosthetic cardiac valves, arterial grafts and arteriovenous malformations. Red cell fragmentation is often visible on a peripheral blood film (Fig. 13A.5).
Microangiopathic haemolytic anaemia (MAHA) refers to fragmentation of red cells due to an abnormal microcirculation. The pathological changes to the microvasculature that cause red cell damage include the deposition of fibrin strands in small vessels, vasculitis, and platelet adherence and aggregation. MAHA develops in conditions such as disseminated intravascular coagulation (DIC). In pregnancy, MAHA may be seen in pre-eclampsia and the associated HELLP syndrome (haemolysis, elevated liver enzymes, low platelets).
Multiple myeloma
Multiple myeloma is a neoplastic proliferation of bone marrow plasma cells. The condition has an annual incidence of approximately 50 per million in the UK, with a median age of presentation of 70 years.
Clinical presentation
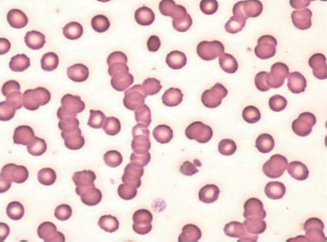
Myeloma may present with a normochromic, normocytic (occasionally macrocytic) anaemia. The ESR is raised, and rouleaux (stacking of red cells on top of each other) is seen on the blood film (Fig. 13A.6). Renal impairment and hypercalcaemia may develop. Osteolytic lesions cause bone pain, and recurrent infections may also be a feature. Occasionally, patients will present with symptoms of hyperviscosity (visual loss, a haemorrhagic tendency, neurological symptoms and cardiac failure), spinal cord compression or have features of amyloidosis.
Figure 13A.5 Red cell fragmentation.
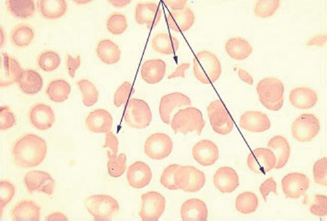
Figure 13A.6 Rouleaux formation.
Investigations
Monoclonal protein is often detectable in serum and/or urine. An IgG paraprotein is detected in approximately two-thirds of cases and an IgA paraprotein in one-third. IgD and IgM myeloma are rare. Normal serum immunoglobulins may be reduced, increasing the risk of infection. Free κ- or λ light chain may be detectable in the urine (Bence–Jones protein).
Rare cases of non-secretory myeloma may be difficult to diagnose as there is no detectable serum or urine paraprotein. Serum free light chain assay is abnormal (showing an abnormal ratio of κ- to λ-light chain in the serum), and a plasma cell infiltrate can be demonstrated in the marrow.
Bone marrow biopsy is required to confirm a clonal plasma cell infiltrate. Skeletal survey (plain X-rays of the skeleton) can demonstrate lytic lesions. CT and MRI are useful for detailed images of the skeleton or soft tissue masses and to investigate suspected spinal cord lesions.
Tables 13A.6 and 13.A7 highlight the diagnostic criteria for asymptomatic and symptomatic multiple myeloma.
Table 13A.6 Diagnostic criteria for myeloma
| Asymptomatic myeloma | Symptomatic myeloma |
| Serum paraprotein >30 g/l and/or bone marrow clonal plasma cells >10% No myeloma-related end-organdamage |
Paraprotein detected in serum and/or urine Bone marrow clonal plasma cells or biopsy-proven plasmacytoma Any myeloma-related end-organ damage |
Table 13A.7 Myeloma-related end-organ damage
|
|
|
|
|
Prognosis
Poor prognostic markers include high serum levels of β-2-microglobulin, low serum albumin and poor risk cytogenetic abnormalities within the neoplastic clone.
Treatment
Prior to commencing chemotherapy, acute renal failure can often be improved with intravenous fluids, although some patients may require renal replacement therapy. Hypercalcaemia also requires intravenous rehydration and occasionally treatment with a bisphosphonate. Urgent radiotherapy is used to treat impending cord compression and can also help to control bone/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


