5
Cardiovascular disorders
5A Introduction to cardiovascular disease
The term cardiovascular disease embraces those conditions affecting the heart and blood vessels. In order to understand these conditions it is necessary to have a basic understanding of the structure and function of the heart and vascular system.
The heart and circulation
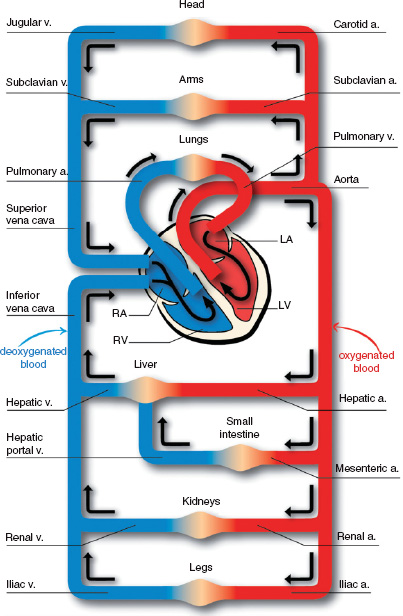
Arteries are thick-walled, high pressure vessels which conduct the blood from the heart to the tissues, while veins are thin-walled, low pressure vessels returning blood to the heart. To complete a full circuit from the left ventricle, oxygenated blood must pass via the systemic arteries to a tissue capillary bed where oxygen is taken up by the tissues. Deoxygenated blood returns via the systemic veins to the right side of the heart, and thence through the pulmonary artery to the pulmonary capillaries, where it is reoxygenated. The pulmonary veins return the blood to the left side of the heart. Figure 5A.1 represents a schematic view of the heart and circulation.
The heart comprises four chambers that pump the blood, with four valves to prevent retrograde flow. The left side of the heart receives oxygenated blood from the lungs, via the pulmonary veins, into the left atrium. Blood flows from here, through the mitral valve, filling the left ventricle, aided by atrial systole (contraction of the left atrium). Ventricular systole ejects blood from the left ventricle through the aortic valve into the aorta, and thence the systemic arterial circulation and capillaries. During ventricular systole, the mitral valve closes to prevent blood flowing backwards into the left atrium, and the aortic valve opens to allow forward flow from the left ventricle.
The right atrium receives deoxygenated blood from the systemic veins via the inferior and superior venae cavae. The blood passes through the tricuspid valve into the right ventricle. Right ventricular systole closes the tricuspid valve and propels the blood through the pulmonary valve into the pulmonary artery, which divides and takes blood to both lungs.
During diastole, the heart muscle relaxes and the mitral and tricuspid valves open to allow ventricular filling, while the aortic and pulmonary valves close to prevent retrograde flow from the aorta and pulmonary artery into the ventricles.
Electrical control
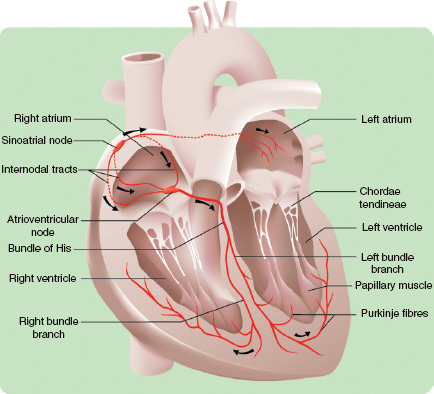
Contraction and relaxation of the cardiac muscle is coordinated by specialised electrical tissues. The sino-atrial node, situated in the right atrium, acts as the pacemaker, which stimulates atrial contraction. This occurs in a wave, with the electrical activity and consequent contraction spreading across both atria towards the atrio-ventricular (A-V) node. The A-V node detects the atrial depolarisation and conducts it, after a short pause, via the bundle of His and the Purkinje fibres to both ventricles. These then depolarise and contract together to eject the blood. A diagram of the cardiac conduction system is given in Figure 5A.2.
Figure 5A.1 The heart and circulation.
Cardiac circulation
The blood supply to the myocardium is carried via the right and left coronary arteries, which branch from the aorta immediately above the aortic valve. These are ‘end arteries’, which means that any blockage cannot be bypassed, and will result in damage to the area of muscle supplied by the blocked artery. Venous blood passes via the coronary veins to the coronary sinus, which runs in the atrio-ventricular groove, and drains directly into the right atrium.
Cardiovascular pathology
Cardiovascular disease (CVD) is the main cause of death in most western societies. For example, in the UK, >1 in 3 people (38%) dies of CVD and it is also one of the main causes of premature death (death before the age of 75 years). The main forms of CVD are coronary heart disease and cerebral vascular disease.
Figure 5A.2 A diagram of the cardiac conducting system.
Atherosclerosis
Atherosclerosis is the most common vascular disease and affects only arteries. It is important in the pathogenesis of both coronary heart disease and cerebral vascular disease.
Definitions
Atheroma is a condition that is characterised by the focal accumulation of lipid in the tunica intima of arteries. The lipid deposits have the texture and appearance of porridge, which is how the condition derives its name (Greek: athere gruel). Fibrosis develops in association with the lipid deposits, resulting in hardening or ‘sclerosis’; hence the term atherosclerosis, which is used synonymously with atheroma.
Risk factors
The cause of atherosclerosis is unknown; however, there are risk factors that are associated with the development of the disease. The risk factors are summarised in Table 5A.1.
The incidence of atherosclerosis increases with age. The incidence in males is much higher than in females up to the age of 55 years. In fact, significant disease is rare in women before the menopause; for this reason it has been proposed that oestrogen has a protective effect on arteries. Following the menopause, the incidence of atherosclerosis in women rapidly increases and then approaches the rates found in males.
Table 5A.1 Risk factors for cardiovascular disease
|
|
|
|
|
|
|
|
|
Cigarette smoking increases the risk of developing the disease and is a particularly strong risk factor in young males. Hypertension and diabetes mellitus are important systemic conditions that predispose to the development of atheroma. Family history is significant because individuals with a first-degree relative that has prematurely suffered the consequences of atherosclerosis are at increased risk of developing the disease.
Raised low-density lipoprotein (LDL)-cholesterol levels in the blood is the most consistent abnormality found in patients with atherosclerosis, although the disease may also develop in those with normal levels of LDL-cholesterol.
Lipid metabolism is a complex process that is influenced by both genetic and environmental factors. Some individuals have a genetic defect in an important lipid receptor, which results in persistently elevated serum levels of LDL-cholesterol with the early development of coronary heart disease, usually by the fifth decade. These observations, in addition to epidemiological data and clinical evidence, led to the development of the ‘diet heart hypothesis’; high dietary fat intake leads to elevated LDL-cholesterol and the development of atheroma. However, the hypothesis is controversial and not all subscribe to this view. In contrast, elevated levels of high-density lipoprotein (HDL) correlate with reduced risk of heart disease. Other, ‘weaker’ risk factors include obesity, lack of exercise and low socio-economic status.
Pathogenesis of atherosclerosis
Atherosclerosis affects medium and large calibre arteries and develops through three morphological stages:
- Fatty streak
- Fibrolipid plaque
- Complicated plaque.
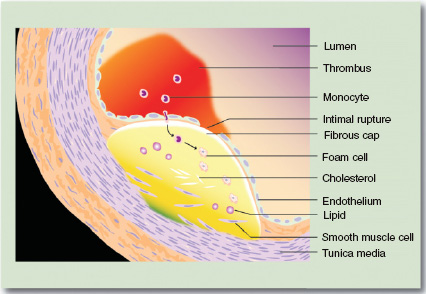
The fatty streak is a subclinical lesion. The fibrolipid plaque is usually silent, but can produce clinical effects if there is significant narrowing of the arterial lumen. Progression to the complicated plaque is asso-ciated with the development of clinically significant disease. The fatty streak is an accumulation of lipid in the intima of the artery wall, just below the endothelial lining of the vessel. The lipid lies free and is also contained within macrophages, so-called foamy macrophages. Fatty streaks have been observed in all age groups, including children.
The fibrolipid plaque is characterised by the deposition of collagen and progressive fibrosis. The lesion has a fibrous cap that bulges into the lumen of the vessel. Beneath the cap there are pools of lipid that contain cholesterol clefts and numerous foamy macrophages. The internal elastic lamina fragments and smooth muscle cells from the tunica media migrate into the lesion and lymphocytes also start to accumulate. In the complicated plaque, the fibrous cap becomes unstable and develops surface defects, referred to as ulcers or intraplaque fissures. This exposes the flowing blood to plaque contents and consequently thrombosis (see later) develops over the plaque (Fig. 5A.3).
The thrombus may partially or completely occlude the vessel lumen. Thrombosis over a plaque will often have serious consequences, especially in narrow vessels such as the coronary arteries, and partially occlusive thrombi are a cause of embolism (see later). The damaged artery may also show progressive calcification. In some cases, damage to the elastic tissue and smooth muscle of the vessel, as a consequence of inflammation, results in gradual permanent dilation of the artery, forming an aneurysm (see later).
Some of the molecular changes that are associated with the development of atherosclerosis have been elucidated. For example, cell adhesion molecules (ICAM-1 and E-selectin) are important in recruiting macrophages into the fatty streak and early fibrolipid plaque. Platelet-derived growth factor (PDGF) is thought to be pivotal in the progression of the lesion, as it stimulates the proliferation of smooth muscle cells and fibroblasts and increases the deposition of collagen, elastin and other matrix proteins within the lesion.
Figure 5A.3 A complicated atheromatous plaque.
Drugs used in the prevention of atherosclerosis
Various drugs are used to reduce plasma lipids. These include the statins such as simvastatin, the fibrates (for example bezafibrate), nicotinic acid, ezetimibe and bile acid sequestrants such as colestyramine.
The statins are the first line treatment. They hinder cholesterol synthesis by inhibiting the enzyme 3-hydroxy-3 methylglutaryl coenzyme A. Statins are of interest to dentists as they can interfere with some drugs that may be used in the management of oral diseases. Myopathy is a side effect of the statins and the chances of this are increased if erythromycin is given to patients taking atorvastatin or simvastatin. Similarly, the antifungal agents ketoconazole and miconazole increase the risk of myopathy in individuals taking simvastatin.
Consequences of atherosclerosis
Atherosclerosis has its main effects on medium to large calibre arteries and is most commonly present in areas of haemodynamic stress, e.g. at arterial bifurcations. The arteries most commonly affected are:
- Coronary arteries
- Cerebral arteries
- Aorta
- Mesenteric arteries
- Iliac and femoral arteries.
The consequences are rather distinct for each named conduit, and the clinical features are dependent on the regions that the blood vessels perfuse. In addition, the precise clinical syndrome depends upon whether the atheroma causes reduction of flow to the organ or tissue or completely occludes the vessel. For example, atherosclerosis of the coronary arteries will cause angina, whereas thrombosis may cause myocardial infarction. Thrombosis over plaques in the vessels at the base of the brain can cause cerebral infarction (stroke).
Thrombosis over atheroma within the aorta can sometimes cause symptoms associated with systemic embolism, but the more common clinical syndrome associated with disease at this site is the abdominal aortic aneurysm. Thrombosis over plaques in the mesenteric vessels may produce small bowel infarction, rupture and subsequent peritonitis. Atheroma of the iliac and femoral vessels causes intermittent claudication (leg pain and weakness brought on by walking) and, when thrombosis develops, gangrene of the lower extremities.
Aneurysms
An aneurysm is a localised permanent abnormal dilatation of a blood vessel due to weakening of the blood vessel wall. The most common aneurysms are those that develop as a consequence of atheroma. Atherosclerotic aneurysms form either at the arch of the aorta, the thoracic aorta or within the abdominal aorta just above the bifurcation of the iliac arteries. The last is the most frequent occurrence and presents with a pulsatile abdominal mass and sometimes lower limb ischaemia. There is also the risk that the aneurysm may rupture and cause a torrential and often fatal retroperitoneal bleed.
The berry aneurysm affects the circle of Willis at the base of the brain. The aneurysm is a small, saccular dilatation that develops at points of branching on the circle of Willis. They typically develop in young hypertensive individuals who have a defect of the muscular wall of the arteries that comprise the circle of Willis. These defects cause areas of weakness, which predisposes to aneurysm formation. Rupture of a berry aneurysm causes subarachnoid haemorrhage.
Other aneurysms are caused by infection such as fungi (mycotic) and syphilitic. Mycotic aneurysms are a consequence of localised infection of an arterial wall. One well recognised source of infection is the embolic material produced during infective endocarditis, which can produce a mycotic aneurysm at the site of impaction, e.g. the cerebral vessels. Rupture of the latter results in cerebral haemorrhage. The syphilitic aneurysm, as the name suggests, is a consequence of chronic infection with Treponema pallidum. The most common site for the syphilitic aneurysm is the root of the aorta where classically aortic ring dilatation and valve incompetence develops. Although commonly recognised in the past, it is now an exceptionally uncommon disease in western society.
Microaneurysms form within capillaries and typically affect cerebral and retinal capillaries. Cerebral micro-aneurysms are usually seen in hypertensive patients, and retinal microaneurysms in diabetics.
The dissecting aneurysm is a ‘false aneurysm’, so called because it is actually a blood-filled space (haematoma) caused by a particular pattern of rupture of the aorta rather than dilatation of the vessel per se. The dissecting aneurysm most commonly commences at the arch of the aorta. Initially there is a small tear in the wall of the aorta and the high pressures generated during cardiac systole forces blood into the defect, causing the tear to propagate. In some instances, the tear may communicate with the lumen of the aorta at some distance from the initial tear, producing a ‘double-barrelled’ aorta. Patients are usually elderly and hypertensive; however, dissecting aneurysms are also seen in patients with connective tissue abnormalities, e.g. Marfan’s syndrome. Patients classically present with severe interscapular back pain, loss of peripheral pulses or with fatal haemopericardium and/or retroperitoneal haemorrhage.
Table 5A.2 The components of thrombosis (Virchow’s triad)
|
|
|
Thrombosis, embolism, ischaemia and infarction
An understanding of these four terms is important for an understanding of CVD. The terms are inextricably linked; a thrombus once formed may undergo embolism, and both thrombosis and embolism cause ischaemia, leading to infarction.
Definitions
- A thrombus is a solid mass of blood constituents formed within the vascular system during life
- An embolus is a mass of material floating free in the vascular system able to become lodged within a vessel and block its lumen
- Ischaemia is an inappropriate reduction in blood supply to an organ or tissue
- Infarction is death of tissue due to ischaemia.
Thrombosis
There are considered to be three factors that predispose to the formation of a thrombus, known as Virchow’s triad, first described by an eminent German pathologist (Table 5A.2).
Virchow postulated that changes in the surface of the vessel, the pattern of blood flow and the blood constituents are important for thrombosis to occur. The presence of all three factors at any one time is not an absolute requirement for thrombosis, however, as thrombosis may occur even if the criterion for only one of the factors is fulfilled.
Thrombosis can occur within the arterial or venous system and within the heart (cardiac thrombosis).
Arterial thrombosis
Arterial thrombosis occurs on atheromatous plaques. Individuals who smoke tobacco have abnormally sticky platelets (change in the blood constituents). The atheromatous plaque protrudes into the lumen of the artery and disturbs the laminar flow of blood, setting up local turbulence (change in the pattern of blood flow). These local currents produce shear forces that can cause endothelial ulceration (change in the wall of the vessel), which leads to platelet aggregation, activation of the coagulation cascade and deposition of fibrin. Subsequently, erythrocytes tend to get trapped in the fibrin meshwork (see Figs 5A.4, 5A.5 and 5A.6). The thrombus develops layer by layer in an incremental fashion; the first layer is composed of platelets with a second layer of fibrin and erythrocytes, then platelets, fibrin and erythrocytes, and so on, forming a lamellated structure.
Figure 5A.4 An atheromatous coronary artery.
Figure 5A.5 Thrombus in a coronary artery.
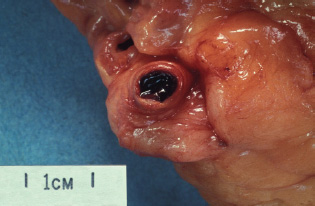
Figure 5A.6 Occluding thrombus in a coronary artery.
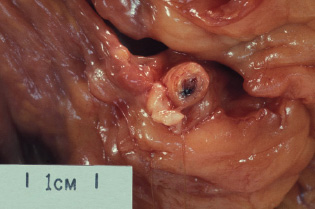
Table 5A.3 Risk factors for deep vein thrombosis
|
|
|
|
|
|
|
|
The incremental layers of the thrombus tend to accumulate on the downstream side of the atheroma and hence the thrombus grows in the direction of the blood flow; a process called propagation.
Venous thrombosis
Venous thrombosis tends to initiate at valves and often develops as a consequence of venous stasis (change in the pattern of blood flow); other predisposing factors include phlebitis (inflammation of a vein). The venous thrombus has the same basic lamellated structure as described for arterial thrombosis, but tends to have a complex pattern of layers, termed a coralline thrombus. Following thrombosis, the vein becomes inflamed, a condition called thrombophlebitis. Venous thrombosis typically develops as a consequence of major trauma and burns, following surgery, cardiac failure, immobility and during pregnancy. The deep veins of the leg are the most common (>90%) site for venous thrombosis (deep vein thrombosis, DVT); the typical clinical signs are a swollen and painful leg. The most clinically important consequence of DVT is a pulmonary embolism. Risk factors for DVT are given in Table 5A.3.
Cardiac thrombosis
Cardiac thrombosis typically affects the chambers of the left side of the heart and occurs on the walls of the heart (mural thrombus) and the heart valves. Atrial thrombosis is caused by atrial fibrillation and mitral valve stenosis. Ventricular thrombosis occurs at the site of myocardial infarction. Valvular thrombus is a feature of rheumatic fever and infective endocarditis.
It is important to appreciate that thrombosis is a dynamic process. In some instances, thrombosis is reversible and the thrombus is effectively broken down by the fibrinolytic pathway, a process called lysis and resolution. In other circumstances, there may be attempts at healing; granulation tissue grows into the thrombus and there is organisation and fibrosis. The latter causes retraction of the thrombus and the patency of the vessel lumen is restored. The vascular buds that comprise granulation tissue can also lead to recanalisation of an occluded vessel. Progressive fibrosis and scarring of thrombus and vessel wall can result in a permanent stenosis (narrowing) of the vessel.
Embolism
An embolus is a mass of material in the vascular system able to become lodged within a vessel and block its lumen. The most common (90%) embolic material is derived from thrombus. Other causes of embolism include atheromatous debris, heart valve vegetations, tumour emboli, fat droplets, amniotic fluid, gas (caisson disease) and foreign material.
In the context of cardiovascular disease, embolism caused by thrombotic debris is the most clinically significant and can have serious consequence for the patient. It may result in sudden death.
Pulmonary embolism
Pulmonary embolism (PE) is the most common clinically recognised embolic event and is usually derived from fragments of a DVT located in the calf or ileofemoral venous segment. Fragments of thrombus become detached from the site of development in the vein and travel through the venous system to reach the right side of the heart. The embolus is then pumped out through the pulmonary arteries to the lungs. The size of the embolus determines the clinical effects. A large embolus may become lodged at the bifurcation of the pulmonary arteries, so called saddle embolus, and cause sudden death. A smaller embolus may lodge in a more peripheral branch of the pulmonary artery resulting in a wedge-shaped infarct of lung tissue; the patient would typically complain of chest pain and breathlessness and sometimes cough up blood (haemoptysis). Rarely, multiple tiny emboli can cause numerous small pulmonary infarctions that may be subclinical, but in some circumstances may lead to pulmonary hypertension.
Systemic embolism
Systemic emboli are mainly derived from thrombus within the left side of the heart or thrombus formed on atheromatous plaques. The clinical consequences are a function of the size and number of emboli and the site where they lodge. Embolism to the cerebral vessels causes cerebral infarction and a transient or permanent neurological deficit (stroke). Smaller emboli may reach the limb extremities and digits causing areas of infarction and gangrene. Other emboli originating in the aorta may lodge in the spleen or the kidney causing segmental infarcts, which are usually asymptomatic. Emboli lodging in the mesenteric vessels may causedeath of large segments of small intestine with bowel perforation and peritonitis.
Ischaemia and infarction
The progression of ischaemia to infarction is determined by a number of factors: vascular anatomy, duration of occlusion, metabolic requirements of the tissue, general circulation, anaemia and the concept of reperfusion injury. If the blood supply to a tissue is predominantly from a single artery and there is little by way of a collateral blood supply, when the main artery becomes occluded ischaemia ensues and progresses to infarction. The duration of vessel occlusion is important because transient ischaemia may not cause tissue damage. Organs with high metabolic demands, e.g. brain and heart, however, are very susceptible to damage even from transient ischaemic events. Poor general tissue perfusion, e.g. in heart failure and anaemia, may compound the effects of localised ischaemia and lower the threshold for tissue infarction.
The degree of tissue damage as a consequence of ischaemia is only fully appreciated following reperfusion of the organ or tissue. This is considered to be due in part to the generation of reactive oxygen species, causing further tissue damage, and bystander tissue damage in the face of the ensuing inflammatory response. This phenomenon is known as reperfusion injury and is one of the complications observed following treatment of ischaemia.
In myocardial infarction (MI) after 6 hours of ischaemia there are no visible morphological changes within the myocardium; however, there are ECG changes and serological evidence of cardiac damage, which form part of the clinical information required to establish a diagnosis of MI. After 24–48 hours the myocardium shows a zone of pallor which has a red rim (inflammation).
Microscopically, the cardiac muscle appears to be structurally intact, but the myocytes show loss of their characteristic muscle fibre striations (coagulative necrosis). After several days the pale dead myocardium becomes soft and is at risk of rupture (haemopericardium; bleeding into the pericardial sac). If the patient survives weeks to months following an MI, the dead tissue is removed by macrophages and there is healing by fibrosis to produce a grey scar.
Infarction in other organs (lung, kidney and spleen) shows similar features to those described for MI and is similarly characterised by coagulative necrosis. In contrast, cerebral infarcts undergo colliquative necrosis. The brain has little by way of robust collagenous supporting tissues, and dead glial tissue and neurons undergo liquefactive degeneration (colliquative necrosis), which is essentially completely cleared by macrophages. As a consequence, a patient who survives cerebral infarction is left with a fluid-filled cystic cavity at the site of injury rather than a fibrous scar.
Clinical features of cardiovascular disease
Symptoms
Chest pain is the cardinal symptom of ischaemic heart disease. In angina pectoris, the coronary arteries, narrowed by atheroma, cannot increase the blood flow to meet the increased demands of the myocardium during exercise. The patient feels a ‘tight’ or ‘heavy’ pain across the chest which commonly radiates to the arms, the neck and jaw. Characteristically it is brought on by exertion and relieved in a few minutes by rest. A differential diagnosis for chest pain is given in Table 5A.4.
When a branch of a coronary artery becomes completely occluded by thrombus, a myocardial infarction (MI) occurs. The chest pain is similar in nature, but more severe and prolonged. It may be accompanied by sweating, nausea, breathlessness or palpitations. It is not relieved by the antianginal drug glyceryl trinitrate (GTN).
Breathlessness, or dyspnoea, is a characteristic feature of heart failure. Depending on the severity, it may occur after only moderate exertion, after only minimal effort or even at rest. Back pressure from the poorly functioning left heart causes congestion of the pulmonary veins and capillaries, which can result in oedema fluid leaking into the alveoli and impairing gas transfer. Because of the changes in hydrostatic pressure with posture, breathlessness may be precipitated by lying down – orthopnoea – or wake the patient from sleep – paroxysmal nocturnal dyspnoea.
Raised pressure in the systemic veins and capillaries in right heart failure causes leakage of oedema fluid into the systemic tissues. The effects of gravity result in swelling of the ankles and legs, often worse in the evenings, and relieved by horizontal posture at night.
The term ‘palpitations’ refers to an abnormal awareness of the heartbeat. It may be a symptom of cardiac arrhythmias. A fast heartbeat may be felt as a ‘fluttering’, or a pause or slow heartbeat be noted as a thump. It may occur normally after exercise and can be a feature of anxiety.
Table 5A.4 A differential diagnosis of chest pain
|
|
|
|
|
|
|
Loss of consciousness due to a lack of blood flow to the brain is termed syncope. Low blood pressure can result in pre-syncope, a feeling of faintness or light-headedness without complete loss of consciousness.
Signs detectable in the clothed patient (see also Chapter 1)
Breathlessness may be detected by observation and measurement of the respiratory rate. Greater than 20 breaths/min at rest is significantly abnormal. Pallor may indicate anaemia or a poor cardiac output, with associated cutaneous vasoconstriction. Under these circumstances, the peripheries may feel cold, and the capillary refill be prolonged. This can be tested by exerting pressure on a fingertip and releasing it. The blanching of the skin produced by the pressure normally resolves in <2 sec as the capillaries refill with blood.
The pulse may be felt in the radial artery at the wrist. Other useful sites are the brachial artery, on the medial side of the arm in front of the elbow (antecubital fossa), and the carotid, below the angle of the jaw. A normal pulse is regular, with a rate between 60 and 100 beats/min, although a resting pulse as low as 40 is found in athletic younger people. Occasional irregularities in an otherwise regular pulse usually indicate ectopic beats, and this finding is seldom of serious significance. A completely irregular pulse usually indicates atrial fibrillation. The strength of volume of the pulse can also give clues to heart disease. It may be weak or thready in states of poor cardiac output of shock. Conversely, it can be strong and ‘bounding’ in states of vasodilation such as thyrotoxicosis or pyrexia due to infection. Furthermore, the strength of an individual beat is dependent on the amount of blood which has filled the heart. Thus, with an irregular pulse such as atrial fibrillation, the beat to beat volume varies, with strong pulses occurring after a pause, and weak pulses where the heartbeat occurs early after the previous beat.
The blood pressure is measured using a sphygmomanometer. Automated sphygmomanometers are now widely available, which give a digital reading of the systolic and diastolic pressures. The blood pressure is taken as described in Chapter 5E. It is usually measured with the patient sitting, at rest, with the arm resting approximately level with the heart.
A raised jugular venous pressure (JVP) is a sign of venous congestion in heart failure. Normally the venous pulsation is visible just above the clavicle, at the root of the neck, in a patient reclining at 45°. In heart failure the vein is distended to a higher level, and may reach the angle of the jaw or above. Chronic elevation of the venous pressure will result in detectable pitting oedema of the ankles. The swelling may be indented by gentle pressure for a few seconds, and the indentation takes a few minutes to disappear.
Peripheral cyanosis, a blue tinge due to deoxygenated blood in the peripheral circulation, may be a sign of peripheral arterial insufficiency, or may occur in states of poor cardiac output such as heart failure or shock. Central cyanosis, best detected in the lips and tongue, indicates reduced oxygenation of the arterial blood leaving the left heart, and is inevitably associated with peripheral cyanosis. Although it may occur in heart failure, it is more commonly seen in lung disease, or congenital malformations of the heart.
Clubbing of the fingers occurs in a variety of conditions, including chronic lung diseases, and inflammatory bowel disease. In cardiac disease it may indicate infective endocarditis, or congenital heart malformations. Splinter haemorrhages (Fig. 5A.7) in the nails are characteristic signs of endocarditis. They appear as small, dark, longitudinal marks under the nail. A few small marks at the nail edge are common due to the trauma or manual activities. In endocarditis, the splinters are greater in number (>5) and away from the edge of the nail. Other rare signs of endocarditis in the hands include Osler’s nodes, which are small, painful nodules in the finger pulp, and Janeway spots, red marks in the palms due to capillaritis.
Full examination of the cardiovascular system includes examination of the heart, lungs and pulses. Auscultation of the heart with a stethoscope may reveal heart murmurs, which are the sounds of turbulent blood flow due to dysfunction of the heart valves. In heart failure, fine crackles or crepitations may be heard at the lung bases. Arterial bruits may be heard over narrowed arter-ies, and are caused by turbulence of the blood passing through the stenosed segment.
Figure 5A.7 Splinter haemorrhages in infective endocarditis. There are usually more than five.
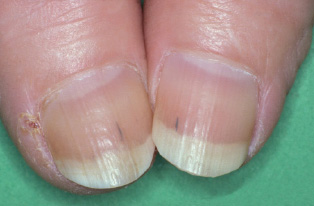
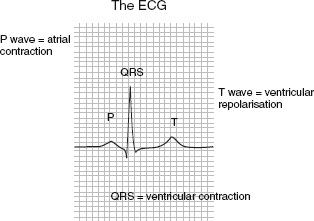
Figure 5A.8 A normal ECG wave form.
Cardiac investigations
The electrocardiogram (ECG) is recorded from skin electrodes placed over the chest and limbs (Fig. 5A.8). It is the definitive test of cardiac rhythm, and can demonstrate changes of both reversible ischaemia in patients with angina, and of acute MI. ECG monitoring during graded exercise on a treadmill is used to diagnose angina, and to give prognostic information in ischaemic heart disease. A summary of some of the more common cardiac investigation methods is given in Table 5A.5.
The chest X-ray may show enlargement of the cardiac shadow in a variety of heart conditions, particularly heart failure. During an episode of acute left-sided heart failure, the pulmonary veins become visibly engorged and oedema fluid in the pulmonary alveoli shows as a diffuse patchy shadowing in both lung fields.
Echocardiography provides real-time ultrasound images of the heart via a transducer moved over the chest wall. More invasive transoesophageal echocardio-grams are used under certain circumstances. The echo-cardiogram shows the structure and function of the valves, the size and contractility of the chambers and can detect fluid in the pericardium.
Cardiac catheterisation involves the insertion of long catheters into the heart, usually via the femoral artery and vein in the groin, under local anaesthetic. These are used to inject X-ray contrast in coronary angiography, and to obtain measurements of intra-cardiac pressure and oxygen saturation to assess the function of the chambers and valves.
Newer imaging techniques such as MRI provide additional information in certain conditions, and can provide some of the information traditionally obtained by cardiac catheterisation in a non-invasive way.
Table 5A.5 Methods for cardiac investigation
|
|
|
|
|
|
Clinical aspects of myocardial infarction
MI is a medical emergency where minutes count. It typically presents with prolonged and severe ischaemic-type chest pain, unresponsive to angina medications. Associated symptoms may include nausea and vomiting, sweating, palpitations, breathlessness and faintness. The patient should be brought by emergency ambulance directly to an emergency chest pain assessment unit or an Accident and Emergency department equipped to give treatment without delay. Causes of chest pain are given in Table 5A.4.
If the diagnosis is confirmed by the ECG showing elevation of the ST segment, the standard treatment is emergency thrombolysis with an intravenous infusion of streptokinase or recombinant tissue plasminogen activator. The aim is to unblock the artery by dissolving the clot, and thereby minimising the damage to the myocardium. Since any delay results in poorer outcomes, there are now protocols whereby thrombolysis may be delivered by trained ambulance staff after confirmation of the ECG result.
Recent research has shown that opening the artery by percutaneous angioplasty, in which a balloon catheter is introduced into the artery during cardiac catheterisation, is superior to thrombolysis. Angioplasty is only available in specialist centres, however, while thrombolysis can be delivered effectively by non-specialist physicians or trained paramedical staff.
Other aspects of treatment of MI involve symptom relief, monitoring for and treating complications, and secondary prevention. Pain in MI is severe, and is treated with intravenous morphine or diamorphine with an antiemetic. The patient has continuous cardiac monitoring to detect arrhythmias or cardiac arrest. Heart failure may occur, and requires intravenous nitrates and diuretics. Echocardiography is used to assess left ventricular function, and ACE inhibitors are started if it is impaired.
All patients are treated with lipid-lowering therapy, β-blockers and antiplatelet drugs. After recovery, the patient’s risk of recurrence of future complications such as heart failure and sudden death is assessed, often using ECG stress testing. This is used to define the need for more invasive treatment such as angioplasty, coronary bypass surgery or implantable defibrillators in those at significant risk of sudden death.
Angina
The diagnosis of angina can often be made on the clinical history, but it is important to differentiate it from other causes of chest pain. Investigations may be required to exclude other causes and to confirm and assess the severity of angina. The key test is the exercise ECG.
Initial treatment is with sublingual GTN, usually as a spray, to be used for rapid relief of symptoms. Several classes of drug are used to prevent attacks, some of which may also affect prognosis. These include β-blockers, calcium channel blockers, longer acting nitrates such as isosorbide mononitrate, and the potassium channel opener nicorandil. Preventive treatment is by risk factor control, with the routine use of lipid-lowering drugs and antiplatelet agents. In severe or resistant cases, cardiac catheterisation is undertaken and arterial stenoses relieved by either angioplasty or open coronary bypass surgery.
5B Heart failure
Introduction
The term heart failure refers to the inability of the heart to pump blood strongly enough to meet the body’s needs. This may result in too little forward flow into the arterial circulation, resulting in low blood pressure and poor tissue perfusion. Conversely, the venous circulation becomes congested with blood, resulting in an increased capillary pressure, which forces fluid from the circulation into the tissues. This increase in extravascular fluid is called oedema.
In physiological terms, heart failure occurs when the heart is unable to pump sufficient blood to supply the body’s demands. The clinical syndrome of heart failure is characterised by a combination of breathlessness, oedema and fatigue. All of these clinical features may be caused by non-cardiac disease, and heart failure may also be aggravated by other concomitant non-cardiac conditions.
Pathophysiology
Three clinically important considerations are:
- What is the nature of the cardiac malfunction?
- What are the body’s adaptive mechanisms?
- What other conditions may be aggravating or precipitating the heart failure?
Cardiac causes of heart failure
Heart failure may result from disease of the heart muscle (myocardium), the valves or disturbances of the heart rhythm.
Heart muscle dysfunction is most commonly due to ischaemic heart disease or hypertension. The myocardium may be damaged by other causes, including drugs and alcohol, nutritional deficiencies and infections. The term cardiomyopathy is used to cover a group of heart muscle disorders of uncertain cause, some of which are inherited.
Myocardial disease commonly results in left ventricular systolic dysfunction, i.e. weakness of the left ventricle, which causes a reduction in the ejection fraction. Normally >60% of the blood in the ventricle is ejected with each beat, and a reduction to 40% or below is associated with a poor prognosis. The term ‘diastolic dysfunction’ refers to poor relaxation of the ventricle, resulting in impaired filling. It is commonly seen in elderly and hypertensive patients, particularly if the left ventricle is hypertrophied, but is less easy to define or diagnose than systolic dysfunction. The strength of cardiac contraction is usually proportional to the degree of stretch or the filling pressure. In myocardial disease, the myocardium is less able to adapt to increased filling pressures.
Stenosis (narrowing) of a valve results in pressure overload of the chamber upstream of that valve. Thus aortic stenosis requires an increased left ventricular pressure to eject the blood, and the ventricle becomes hypertrophied and can eventually fail. Mitral stenosis results in a raised left atrial pressure, resulting in dilation of the left atrium.
Valvular regurgitation results in volume overload. For example, in aortic regurgitation, a proportion of the ejected blood returns to the left ventricle from the aorta, and has to be pumped out a second time.
Arrhythmias can precipitate heart failure in an otherwise normal heart. A severe tachycardia impairs cardiac filling during the shortened diastole, while a severe bradycardia reduces the number of contractions to maintain cardiac output. The most common chronic arrhythmia is atrial fibrillation, which also impairs left ventricular filling due to the loss of atrial contraction.
Neurohormonal adaptive responses
The circulatory consequences of heart failure, for whatever cause, are:
- Reduced cardiac output
- Increased intracardiac pressure
- Increased back pressure in the venous circulation, resulting in increased capillary pressure and consequent leakage of fluid into the tissues (oedema).
Homeostatic mechanisms that have evolved to maintain fluid balance and blood pressure in states such as haem-orrhage, dehydration or fluid overload then come into play.
Reduced cardiac output results in activation of the sympathetic nervous system and the renin–angiotensin– aldosterone system, in an attempt to maintain the blood pressure and cardiac output. Release of the catechol-amines adrenaline and noradrenaline stimulates both the force and rate of contraction of the heart, and constricts the blood vessels to maintain the blood pressure. Angiotensin is also a vasoconstrictor, while aldosterone stimulates sodium retention in the kidney, and consequently water retention, increasing the circulating volume. Conversely, stimulation of stretch receptors in the atria and ventricles causes release of natriuretic peptides which stimulate salt and water loss via the kidneys.
While many of these mechanisms may be helpful in the short term in acute heart failure, they are maladaptive in chronic heart failure. Vasoconstriction, although maintaining the blood pressure, increases the resistance against which the failing heart has to pump. Sodium and water retention aggravates oedema, and increases the circulating volume, thereby stretching the heart further. Constant stimulation of the myocardium by catecholamines results in further myocardial damage.
Exacerbating conditions
A number of non-cardiac conditions can aggravate heart failure. Indeed, clinical heart failure can occur in a normal heart if it is under sufficient external stress.
- Anaemia and hypoxia increase the cardiac workload
- Arterio-venous shunts, including those created for haemodialysis in renal patients, require an increased cardiac output
- Hyperthyroidism causes sympathetic stimulation and concomitant peripheral vasodilation, resulting in a tachycardia and high cardiac output
- Severe renal failure impairs excretion of salt and water, resulting in fluid overload
- Excessive intravenous fluid therapy can precipitate pulmonary oedema, even with a normal heart.
Clinical aspects of heart failure
The clinical symptoms and signs of heart failure are described in Chapter 5A. These may vary between patients. Left-sided heart failure will cause predominantly breathlessness due to pulmonary congestion, while right-sided heart failure causes systemic venous congestion, with a raised venous pressure and pitting oedema. The two commonly co-exist.
Clinical assessment should answer the following questions:
- Is this really heart failure?
- What is the underlying cardiac problem?
- Are there any exacerbating conditions?
- What treatment is required – initially to relieve symptoms and ultimately to improve prognosis?
Some cases of apparent heart failure are due to other conditions. Lung disease is a common cause of breathlessness, and often co-exists with ischaemic heart disease, particularly in smokers. Ankle oedema may be due to varicose veins, DVT, immobility, obesity or a decreased serum albumin concentration.
The cardiac diagnosis may be apparent clinically, but all patients should have an ECG, chest X ray (Fig. 5B.1) and an echocardiogram. The latter gives the most detailed information about the function of the cardiac chambers and valves.
Anaemia, renal failure and thyrotoxicosis may all aggravate heart failure, and a full blood count, urea and electrolytes and thyroid function tests should be performed. In ischaemic heart disease, risk factors should be assessed and treated.
Figure 5B.1 A patient with heart failure. Pulmonary oedema is shown, with an increase in lung markings and increased heart size.
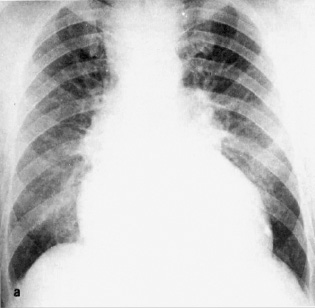
Treatment of heart failure
Acute pulmonary oedema is a medical emergency requiring hospital admission. The immediate ‘first aid’ treatment is to sit the patient up to optimise both the haemodynamic stress on the heart and the mechanics of breathing. Oxygen should be given, and patients with ischaemic heart disease should take their GTN sublingual preparation.
In hospital, an ECG is taken to assess cardiac causes such as an acute arrhythmia or MI. Blood tests are sent, and a chest X-ray performed. Intravenous furosemide is given, which acts both as a vasodilator, reducing cardiac workload, and as a diuretic, reducing circulating volume. An intravenous nitrate infusion can be given, and morphine or diamorphine provides symptomatic relief from breathlessness. Further management depends on the underlying cause.
Chronic heart failure is assessed and treated similarly, initially with oral loop diuretics to relieve fluid overload. When the heart failure is due to left ventricular systolic dysfunction, with a low ejection fraction on the echocardiogram, a number of medications acting on the neurohormonal pathways described above have been shown to reduce both morbidity and mortality.
Drugs used to manage heart failure
The drugs used to treat heart failure are shown in Table 5B.1. Most of the drugs are described more fully in other parts of the chapter.
- ACE inhibitors reduce mortality by about 25% at all levels of severity. They have to be started at a low dose and titrated upwards with monitoring of the blood pressure, urea and electrolytes, as they can cause renal impairment and hyperkalaemia.
- Angiotensin receptor blockers act in a similar way.
- Low doses of spironolactone, an aldosterone antagonist, reduce mortality in severe heart failure already treated with ACE inhibitors.
- The β-blockers carvedilol and bisoprolol reduce mortality by >30%. β-blockade can aggravate acute heart failure, since acute adrenergic stimulation increases the contractility of the heart, and these drugs are introduced slowly once symptoms and fluid overload are controlled by diuretics.
- Phosphodiesterase inhibitors include enoximone and milrinone. Inhibition of the enzyme phosphodiesterase causes intracellular accumulation of cAMP, which results in a rise in calcium entry. This increases the force of myocardial contraction.
Table 5B.1 Drugs used to treat heart failure
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


