Medically compromised children
Kerrod B Hallett, Sherene Alexander, Meredith Wilson, Craig Munns, Angus C Cameron and Richard P Widmer

Introduction
Comprehensive dental care of a medically compromised child requires consideration of their underlying systemic condition and coordination of their dental treatment with their medical consultant. Although dental problems are common in this group, their oral health is overlooked frequently by the medical profession. The term used to identify this particular group, ‘medically compromised children’, has been replaced recently by the more general term ‘children with special needs’. However, the older term is still relevant because it reminds the dentist that these children often have medical conditions that can affect dental treatment or that they can present with specific oral manifestations of a systemic disease. This chapter discusses the common paediatric medical conditions that require consideration in the provision of optimal dental treatment. The prevention of oral diseases is important in children with chronic medical problems (Figure 12.1), as oral complications can severely compromise a child’s medical management and overall prognosis.

Cardiology
Congenital heart disease
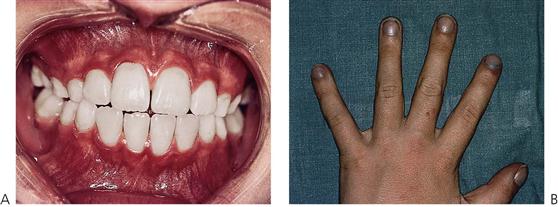
Congenital heart disease (CHD) has an incidence of approximately 8–10 cases per 1000 live births and represents the largest group of paediatric cardiovascular diseases. Although most lesions occur individually, several form major components of syndromes or chromosomal disorders such as Down syndrome (trisomy 21) (see Figure 12.2A) and Turner syndrome (45, X chromosome) with over 40% of children being affected. However, in the majority of cases, no cause can be determined and a multifactorial aetiology is often assumed. Known risk factors associated with CHD include maternal rubella, diabetes, alcoholism, irradiation and drugs such as thalidomide, phenytoin sodium (Dilantin) and warfarin sodium (Coumadin). Turbulent blood flow is caused by structural abnormalities of the heart anatomy and presents clinically as an audible murmur. The degree of clinical morbidity is determined by the haemodynamics of the lesion. Congenital heart disease can be classified into acyanotic (shunt or stenotic) and cyanotic lesions depending on clinical presentation. Eight common conditions account for 85% of all cases.
Acyanotic conditions
The acyanotic group of conditions is characterized by a connection between the systemic and pulmonary circulations or a stenosis (narrowing) of either circulation. Infants often present with feeding difficulties, breathlessness and failure to thrive. Shunts are from the left to right. The most common anomalies and their specific sites are:
• Atrial septal defect (ASD) – usually located near foramen ovale.
• Ventricular septal defect (VSD) – in the membranous septum of the ventricular wall.
Acyanotic defects with obstruction include:
Cyanotic conditions (Figure 12.2)
All cyanotic conditions exhibit right-to-left shunting of desaturated blood. Cyanotic defects become clinically evident when 50 g/L of desaturated haemoglobin is present in peripheral arterial blood. Infants with mild cyanosis may be pink at rest but become very blue during crying or physical exertion. Children with cyanotic defects are at significant risk for desaturation during general anaesthesia and preoperative consultation with the paediatric cardiologist and anaesthetist is essential.
The most common cyanotic lesions are:
Other cardiovascular diseases
Other common paediatric cardiovascular disorders include cardiomyopathies such as myocardial disease and pericardial disease, cardiac arrhythmia, infective endocarditis and rheumatic heart disease (RHD). Both CHD and RHD can predispose the internal lining of the heart to bacterial or fungal infection (infective endocarditis) and lead to the formation of friable vegetations of blood cells and organisms. Vegetations may embolize and cause renal, pulmonary or myocardial infarcts or cerebrovascular accidents. Streptococcus viridans, a common commensal organism in the oral cavity, is most frequently responsible for chronic infective endocarditis, whereas Staphylococcus aureus is often implicated in the acute fulminating form of infective endocarditis.
Dental management
Several important principles need to be followed when managing children with cardiac disease. Transient bacteraemia can occur following invasive dental procedures and potentially cause infective endocarditis in a susceptible patient. Therefore, all children with CHD or previous RHD require antibiotic prophylaxis to reduce the risk of infective endocarditis (see Appendix E). Those children who have been previously taking long-term antibiotics should be prescribed an alternative medication as per the protocol to avoid development of resistant oral organisms. In addition, a preoperative oral antiseptic mouthwash, such as 0.2% chlorhexidine gluconate, is recommended to reduce the oral bacterial counts.
Children with CHD have a higher prevalence of enamel anomalies in the primary dentition and concomitant risk of early childhood caries. Some cardiac medications may contain up to 30% sucrose and dietary prescription with high-caloric supplements (Polyjoule) further potentiate caries risk. Meticulous oral hygiene and preventive dental care, such as fissure sealants and topical fluoride therapy is recommended to reduce the risk of dental caries in susceptible children.
Dental disease in children with cardiac disorders can seriously complicate their medical management. Children with advanced cardiovascular disease should receive only palliative dental care until their medical condition has been stabilized. Aggressive treatment of pulpally involved primary teeth is recommended. Pulpotomy or pulpectomy is contraindicated in these children due to the possibility of subsequent chronic bacteraemia. Although routine treatment in the dental surgery environment is possible, it is often preferable to manage children with multiple carious teeth under general anaesthesia in the hospital environment. This protocol allows completion of treatment with one invasive procedure and negates the risk of infective endocarditis with further operative procedures. If multiple visits are planned, there is a need to prescribe alternative antibiotics or wait for a month between appointments to reduce bacterial resistance.
A thorough preoperative assessment of the child’s regular medication (including anticoagulants, antiarrhythmics, and antihypertensives) is essential to avoid any potential drug interactions during treatment. There is no contraindication to the use of vasoconstrictors in local anaesthetic solutions. If conscious sedation is used, vital signs and oxygen saturation during the procedure should be carefully monitored. Avoid the use of electrosurgery, electronic pulp testers and ultrasonic cleaning devices in children with cardiac pacemakers, in case of potential interference. Some common impediments are non-compliance with oral hygiene and dietary advice, postoperative infection and bleeding.
Haematology
Disorders of haemostasis
Primary haemostasis is initiated after injury to a blood vessel with the formation of a primary platelet plug. This process is mediated by interactions between the platelets and coagulation factors in the plasma and the vessel wall. Secondary haemostasis or coagulation is also triggered by the initial injury and reaches its greatest intensity after the primary platelet plug is formed. Fibrin deposition provides the framework for the formation of a stable blood clot.
Prolonged bleeding can occur when either phase of haemostasis is disturbed. The clinical manifestations of a haemostasis disorder vary depending on the phase affected. Defects in primary haemostasis generally result in bleeding from the skin or mucosal surfaces, with the development of petechiae and purpura (ecchymoses). These disorders include von Willebrand’s disease as well as defects in platelet function. In contrast, defects in secondary haemostasis, such as haemophilia, lead to bleeding that tends to be more deep-seated in muscles and joints. In both disorders, uncontrolled prolonged oral bleeding can occur from innocuous insults such as a tongue laceration or cheek biting.
Children with haemostasis disorders can be identified from a thorough medical history, examination and laboratory tests. Questions should reveal episodes of spontaneous bleeding or bruising; the occurrence of prolonged bleeding in other family members and prescription of anticoagulant medication. A physical examination of skin (unusual areas of bruising on the chest or back or bruising from lying on a toy), joints and oral mucosa should be undertaken for evidence of petechiae, ecchymoses and haematoma. If a haemostasis disorder is suspected, referral to a haematologist is recommended for evaluation and laboratory blood tests.
Laboratory tests
Classification
Vascular disorders
Vascular disorders are characterized by increased capillary fragility and include the purpuras, hereditary haemorrhagic telangiectasia, haemangiomas, vitamin C deficiency, Henoch Schönlein purpura and connective tissue disorders such as Ehlers–Danlos syndrome.
Platelet disorders
Platelet disorders can be either a deficiency (thrombocytopenia) or dysfunction.
Thrombocytopenia
Thrombocytopenia is defined as a platelet count <150 × 109/L. Clinical signs and symptoms associated with decreased platelet counts are as follows:
• <75 × 109/L – May exhibit post-surgical haemorrhage.
• <25 × 109/L – Spontaneous haemorrhage, easy bruising.
• <15 × 109/L – Petechiae appear on the skin.
• <5 × 109/L – Oral petechiae, submucosal and mucosal bleeding.
Thrombocytopenia may occur as an isolated entity of unknown cause (idiopathic thrombocytopenic purpura, ITP), as a result of marrow suppression by drugs or from other haematological diseases such as aplastic anaemia. Marrow replacement by neoplastic cells in haematological malignancies will also result in thrombocytopenia. Children undergoing chemotherapy will have decreased platelet counts.
Thrombocytosis
Thrombocytosis is an increased number of platelets (>500 × 109/L) and may be associated with prolonged bleeding due to abnormal platelet function. Myeloproliferative disorders may present with thrombocytosis.
Platelet function disorders
These may be congenital or acquired. The most common cause of acquired platelet dysfunction is the use of non-steroidal anti-inflammatory drugs (e.g. aspirin). Administration of cyclo-oxygenase inhibitors will result in blockage of the production of thromboxane A2 for the life of the platelet (7–9 days). This results in a decrease in platelet aggregation. Some metabolic diseases such as Gaucher’s disease also manifest as defects of platelet function.
A decrease in the number of platelets or platelet dysfunction will result in failure of initial clot formation. Children with thrombocytopenia will bleed immediately after trauma or surgery, unlike those with haemophilia, who usually start to bleed 4 h after the incident. The most common oral manifestations are petechiae and ecchymoses. There may also be spontaneous gingival bleeding and prolonged episodes of bleeding after minor trauma or tooth brushing.
Inherited coagulation disorders
Coagulation disorders result from a decrease in the amount of particular plasma factors in the coagulation cascade. The most common disorders are haemophilia A and von Willebrand’s disease, both manifesting a decrease in factor VIII levels. The factor VIII is produced by endothelial cells and is composed of two portions. The largest part of the molecule is the von Willebrand’s factor and is responsible for initial platelet aggregation. The factor VIII part of the complex and factor IX are responsible for activation of factor X in the intrinsic pathway of the coagulation cascade. Other disorders of coagulation include vitamin K deficiency, liver disease and disseminated intravascular coagulation usually from overwhelming (Gram-negative) infection.
Coagulation disorders are classified according to the defective plasma factor; the most common conditions are factor VIII (haemophilia A) and factor IX (haemophilia B or Christmas disease). von Willebrand’s disease occupies a unique position in that both platelet and factor VIII activity is decreased, therefore both bleeding time and APTT are prolonged.
Haemophilia A
This is inherited as an X-linked recessive disorder with deficiency of factor VIII, and occurs 1 in 10 000 live male births. Spontaneous mutation occurs in 30% of cases. The disease is classified as:
• Severe (<1% factor VIII) – with spontaneous bleeding into joints and muscles.
• Moderate (2–5% factor VIII) – with less severe bleeding usually following minor trauma.
Factor VIII assay is usually performed after the initial diagnosis of a coagulopathy. Affected children and their families require considerable medical support and may have an indwelling central line for regular factor VIII concentrate infusion.
Haemophilia B or Christmas disease
This disease has clinical features similar to factor VIII deficiency. It is also inherited as an X-linked recessive gene and results in prolongation of the APTT. It is diagnosed by specific assay of factor IX.
von Willebrand’s disease
von Willebrand’s disease is inherited as an autosomal dominant trait (gene locus 12p13). The most common clinical manifestations include epistaxis and gingival and gastrointestinal bleeding. von Willebrand factor is found in the plasma, platelets, megakaryocytes and endothelial cells and circulates as a major component of the factor VIII molecule complex. This disease is divided into various subtypes, based on the platelet and plasma multimeric structure of the von Willebrand factor.
Dental management
Dental management of children with suspected haemostasis disorders should begin with screening laboratory tests. If tests are abnormal, haematological consultation is required for a definitive diagnosis. Invasive dental procedures should be performed only after the extent of the problem has been determined. Extractions must never be performed without first consulting the haematologist. It is preferable to have platelet levels >80 × 109/L before extractions. Endodontic procedures may be preferable to extractions in order to avoid the need for platelet transfusion.
Medical management
Haemophilia B
von Willebrand’s disease
Other factor replacements
1-Deamino (8-D-arginine) vasopressin (DDAVP)
Post-surgical administration of antifibrinolytic agents such as tranexamic acid (Cyklokapron) 25 mg/kg loading dose and 15–20 mg/kg three times daily for 5–7 days is helpful in preventing clot lysis. During the time that antifibrinolytics are given, the parent and child should be instructed not to use straws, metal utensils, pacifiers or baby bottle teats.
Characteristically, haemophilia bleeds are delayed 12–24 h, as primary haemostasis is not impaired, and local pressure has little effect. It is worth noting that mild haemophilia can go undiagnosed. The APTT is not sensitive to detect mild deficiencies of FVIIIc and levels of FVIIIc 25–30 IU/dL can be associated with a normal APTT. In addition, FVIIIc values in mild haemophilia are temporarily increased (as occurs in unaffected persons) by stress, exercise and bleeding. If there is a convincing history of a bleeding tendency always do a specific factor assay even if the initial screening tests are normal.
The normal regimen for DDAVP is 0.3 µg/kg intravenous infusion over 1 h before surgery followed by tranexamic acid 15–20 mg/kg orally every 8 h for 7 days. After 9–12 h, if the FVIIIc levels are still low (50–60%), then the original dose of DDAVP may be repeated. If repeated doses are planned or required it is important to fluid restrict the patient and monitor electrolytes. Repeated doses of DDAVP may cause fluid retention and hyponatraemia. This regimen is useful in von Willebrand’s disease and children on renal dialysis.
Clinical Hints
Questions commonly asked by parents are:
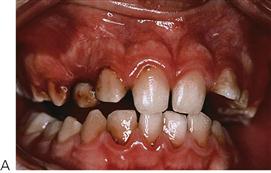
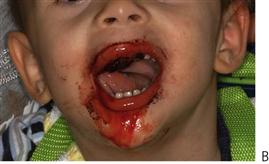
• Will my child’s teeth fall out normally? Usually yes, unless continually traumatized, there is normally no abnormal bleeding associated with exfoliating primary teeth. However, if there is prolonged mobility and oozing occurs, then extraction may be necessary under appropriate factor cover to reduce the risk of persistent bleeding (Figure 12.3).
Anticoagulant therapy
Management of children on anticoagulant therapy needs special consideration. Anticoagulants are usually prescribed for children with valvular heart disease and prosthetic valves to reduce the risk of remobilization. If extractions or surgery are required, it is necessary to decrease the clotting times to facilitate adequate coagulation but not to such an extent so as to cause emboli or clotting around the valves. The dental management of these children is also complicated by their congenital cardiac defect and antibiotics are required for prophylaxis against infective endocarditis.
Therapeutic drugs used
• Oral warfarin sodium (Coumadin):
• Vitamin K antagonist depleting factors II, VII, IX and X.
• Shorter acting and has an immediate onset (inhibits factors IX, X and XII).
• Enoxaparin sodium (Clexane):
• Low-molecular-weight heparin which inhibits factor Xa and thrombin.
Children on anticoagulant therapy should stop taking warfarin 3–5 days prior to the surgery date. In those in whom there is a significant risk for thrombosis with sub-therapeutic warfarin level, parenteral anticoagulation may be necessary. This is generally achieved with enoxaparin sodium (Clexane) 1.5 mg/kg subcutaneously once daily (mane) via Insuflon. This drug is omitted on the morning of surgery. With the use of this regimen, the child may be admitted to hospital on the day of dental surgery. Warfarin is recommenced in normal dose on the evening of surgery. If further enoxaparin sodium prophylaxis is required, it should be given the morning after surgery and continued until the PT and international normalized ratio (INR) are therapeutic. Monitoring of enoxaparin sodium is rarely required. In emergency situations with prolonged bleeding from oral wounds post-surgery, following recommencement of warfarin, FFP (fresh frozen plasma) may also be of benefit.
Local haemostatic measures
Management of oral haemorrhage
Unexpected bleeding from the oral cavity can occur at any time. There may have been a slow ooze for several days or, in the other extreme, there may be a significant sudden oral bleed. Such bleeding can occur without warning and may not be associated with any prior investigative or operative work. As well, haemorrhage from the mouth can occur following such routine procedures as biopsy, restorative work or tooth extraction.
The initial management of such cases involves identifying the exact site of haemorrhage, controlling the bleeding and then preventing a recurrence. In the cases of haemorrhage from the mouth that has not been associated with any dental procedure, clinicians should take an accurate history of the bleeding, the duration, lost volume and any causative factors. Abnormal bleeding may occur around an erupting tooth, from an exfoliating tooth site or may be associated with physical and sexual child abuse or congenital vascular anomalies such as arteriovenous malformations. The possibility of a childhood malignancy should also be considered.
In cases of oral haemorrhage following dental procedures, the following steps should be taken (it is important to prevent or minimize bleeding in the first instance):
• A sensible limitation of surgical trauma.
• Digital compression of the alveolus after tooth extraction.
• Packing of the socket with a resorbable gel.
• Adequate suturing of extraction sites to help reduce postoperative complications.
• Pressure application to the surgical site with gauze packs.
• Construction of a removable splint is recommended following more extensive surgery.
• Prescription of non-aspirin medication are necessary to avoid any parent misunderstanding.
In cases of severe uncontrollable haemorrhage following tooth extraction that can occur due to arteriovenous malformations, remember that the best method of controlling the bleeding is to replant the extracted tooth back into the socket and suture it well.
Red cell disorders
Anaemia
Anaemia is considered to be present if the haemoglobin level falls below 100 g/L. The cause of anaemia in children may be due to blood loss, iron, folate and vitamin B12 deficiency, bone marrow failure, haemolysis of red blood cells or anaemia of chronic disorders. It is usually an incidental finding in the routine dental management of children. A full blood count (FBC) is usually ordered when children present with pallor, lethargy, fever, bruising, undiagnosed systemic or oral pathology after major trauma associated with excessive blood loss or on work-up before surgery for other medical conditions. When unexpected anaemia is discovered, follow-up by the paediatrician is required.
Haemolytic anaemia
Acute haemolytic disease of the newborn or erythroblastosis fetalis is caused by ABO incompatibility and Rhesus (Rh) iso-immunization. There will be discolouration of those primary teeth that are calcifying at the time of birth. The cusp tips of the first permanent molars may also be affected. A yellow-green staining is most commonly seen as a result of high levels of unconjugated bilirubin.
Glucose 6-phosphate dehydrogenase (G6PD)
G6PD deficiency also results in acute haemolytic anaemia when the child is exposed to certain drugs (sulphonamides, chloramphenicol, aspirin, antimalarials) or infection (hepatitis).
Aplastic anaemia
Aplastic anaemia is defined as a decrease or absence of haemopoiesis in the bone marrow that is not due to marrow involvement or recognized disease process. FBC and bone marrow aspirate confirms the diagnosis. Bone marrow transplantation is the treatment of choice for moderate to severe aplastic anaemia.
Haemoglobinopathies
Thalassaemia
The haemoglobinopathies are a group of genetic disorders involving the globin chains of the haemoglobin (Hb) complex. These diseases comprise two main groups: the structural haemoglobinopathies, resulting in abnormal globins (HbE, HbS) and the thalassaemias. The thalassaemias represent a group of autosomal recessive disorders, common in patients from the Mediterranean, North Africa, the Middle East, India and Central Asia, expressing mutations of genes responsible for the production of any of the haemoglobin chains.
Haemoglobin is a tetrameric protein comprising four globin protein subunits. Adult blood contains haemoglobin A (HbA), comprised of two α-chains and two β-chains and a small amount of haemoglobin A2 (HbA2) comprised of two α-chains and two δ-chains. Children also produce fetal haemoglobin (HbF – two α-chains and two γ-chains), which has a much higher oxygen affinity. Fetal haemoglobin levels decrease after 6 months of age from around 70% at birth to trace amounts in adulthood.
α-Thalassaemia – is caused by deletions or mutations of the four alpha globin genes on chromosome 16. One to four genes may be affected, resulting in a relative overproduction of β-chains. Homozygous α-zero thalassaemia (four alpha globin genes deleted) is incompatible with life, while carriers (1–2 gene deletions) have no clinical symptoms. Children with HbH disease (three alpha genes deleted/abnormal) may have mild anaemia or a transfusion dependent anaemia.
β-Thalassaemia – Of more clinical significance is homozygous β-thalassaemia major (Cooley’s anaemia). Due to the absence of the β-chain, there is a compensatory increased production of HbA2 and HbF. As erythropoiesis is inadequate, the bone marrow is reactive and there is compensatory intermedullary haemopoiesis in the maxilla and diploe of the skull. There may be severe haemolytic anaemia with marked hepatosplenomegaly and failure to thrive. Those children with sickle/β-thalassaemia show evidence of vascular thrombosis with ischaemia to organs, especially bones.
Due to maxillary and zygomatic overgrowth there is often a severe Class II Division 1 malocclusion with separation of teeth and widening of the periodontal ligament space. Lateral skull radiographs demonstrate a ‘hair on end’ appearance. Children are given regular packed red cell hypertransfusions until the haemoglobin rises to 140–150 g/L and desferrioxamine, an iron-chelating agent, to increase iron excretion. When excessive haemosiderosis in the spleen adds significantly to the haemolysis rate, elective splenectomy is performed.
Sickle cell disease
This is different from the other haemoglobinopathies in that the red blood cells are more susceptible to haemolysis and have difficulty passing through small blood vessels causing infarcts and ischaemia of organs and bone. These patients are usually asymptomatic unless subjected to low oxygen concentrations and this may be an issue when a general anaesthetic is required. Blood transfusions, analgesics, antimicrobials, adequate hydration and other life-supportive measures are necessary.
Dental management
Consultation with the child’s haematologist prior to treatment is essential to arrange haematological preparation and transfusion. It is important to schedule dental treatment shortly after blood transfusions and provide antibiotic prophylaxis, especially if the child has had splenectomy. Avoid elective treatment if haemoglobin level is <100 g/L. Minimize stress that might compromise the child’s ability to oxygenate the tissue adequately. Respiratory depressants should be avoided and additional oxygenation during conscious sedation or general anaesthesia is desirable along with the use of pulse oximetry. Local anaesthesia is not contraindicated but the use of prilocaine (Citanest) is not advised due to the formation of methaemoglobin. Vasoconstrictors in the standard dose are not contraindicated. Orthodontic treatment may be undertaken but teeth will move quickly through the bone and relapse will most likely occur.
Immunodeficiency
Immunodeficiency may be caused by quantitative or qualitative defects in neutrophils, primary immunodeficiencies, involving T cells, B cells, complement or combined defects and secondary immunodeficiency or acquired disorders.
Qualitative neutrophil disorders
Chemotactic disorders
Quantitative neutrophil disorders
Neutropenia
This is defined as <1.8 × 109 cells/L. Life-threatening sepsis is associated with a level of neutrophils <0.5 × 109 cells/L. Neutropenia can occur in the following situations:
• Infiltration of bone marrow by neoplastic cells.
• After administration of cytotoxic drugs used for treatment of childhood malignancy.
• Cyclic neutropenia (21–28 day cycling).
• Nutritional: protein-calorie malnutrition, vitamin B12 deficiency, copper deficiency.
• Pseudo neutropenia: usually mild and spontaneously resolves.
Primary immunodeficiencies
Secondary or acquired immunodeficiencies
Secondary or acquired immunodeficiencies include those conditions acquired during childhood, such as:
• Human immunodeficiency virus (HIV) infection.
• Drug-induced immunodeficiency (cytotoxics, corticosteroids, cyclosporin A, tacrolimus).
These can also occur in children who have undergone bone marrow transplantation and radiotherapy (radiotherapy-induced immunodeficiency).
Combined immunodeficiencies
Dental implications
Both neutrophil and T-cell-mediated immunodeficiencies predispose the child to infection by compromising the host defence system. Opportunistic organisms that do not usually cause disease in a healthy child can proliferate in the oral cavity of the immunodeficient host. Common oral manifestations seen are:
• Acute pseudomembranous candidiasis (Figure 12.5B).
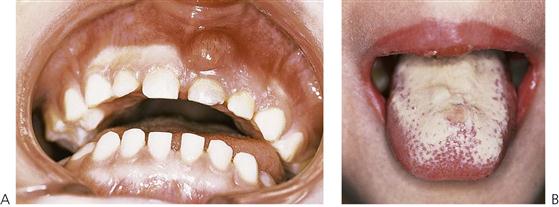
• Generalized prepubertal periodontitis (Figure 12.4).
• Recurrent aphthous ulceration.
Generally, B-cell deficiencies exhibit fewer oral complications but are often associated with chronic bacterial infections such as pneumonia, otitis media and skin lesions.
Dental management
Regular review of the developing dentition, gingivae and mucosa and the institution of a preventive programme are essential for maintenance of healthy hard and soft tissues. Elimination of any potential oral focus of infection during the course of medical treatment is the primary objective.
The underlying deficiency must be fully assessed and the likelihood of oral complications endangering the child’s medical status should be evaluated. An individual risk–benefit assessment of any oral lesion must be considered with regard to the overall management plan.
A decision whether to extract or maintain carious teeth and exfoliating primary teeth must be based on the worst case scenario during the immunodeficiency period. If a carious lesion cannot be stabilized with an adequate interim restoration, then extraction is the preferred treatment. In a case being prepared for bone marrow transplantation, all mobile primary teeth should be removed at least 2 weeks prior to the conditioning phase.
Thorough dental scaling and prophylaxis and the provision of custom trays for delivery of medication (antiseptic or fluoride gels) prior to commencement of head and neck radiotherapy is also recommended to prevent oral sepsis and radiation-induced dental caries.
Prophylactic antimicrobials specific for commensal oral organisms determined from culture and sensitivity tests are indicated during the course of medical treatment. Biopsy specimens may assist the diagnostic process. The antimicrobial protocol may include appropriate antibiotics (amoxicillin trihydrate and ampicillin, vancomycin), acyclovir sodium if HSV-positive, ganciclovir if cytomegalovirus-positive, antifungals (topical nystatin and amphotericin B) and twice daily 0.2% chlorhexidine gluconate (Curasept) mouthwashes during the active therapy phase.
Acquired immunodeficiency syndrome (AIDS)/HIV
HIV infection has been identified in increasing numbers of children with otherwise unexplained immune deficiency and opportunistic infections of the type found in adults with acquired immune deficiency syndrome (AIDS). For the limited purposes of epidemiological surveillance, the Centers for Disease Control (CDC) characterizes a case of paediatric HIV infection as a reliably diagnosed disease in children that is at least moderately indicative of underlying cellular immunodeficiency, and with which no known cause of underlying cellular immunodeficiency or any other reduced resistance is reported to be associated.
Transmission
The main transmission media are body fluid, such as blood and semen. Saliva contains low and inconsistent levels of the HIV virus and is unlikely to provide a significant mode of transmission. Consequently, the two major routes of transmission in children are vertical (from an infected mother) and from blood products, with children with haemophilia being most at risk. Vertical transmission rates are up to 39% and occur before, during or after birth. Infection from breast-feeding may be up to 29%.
Risk factors
Serodiagnosis and immune function
The screening ELISA test for HIV antibodies is liable to give false negatives and any apparently positive results must be confirmed by the western blot assay. Antigen assays are far more reliable, but a failure to detect virus or antigen in a young antibody-positive child does not exclude infection. A positive virus or antigen test is likely to indicate infection. Due to the long incubation period and the limitations of medical history and serodiagnosis, it must be assumed that all blood derivatives may be infectious.
The human immunodeficiency virus attaches to the CD4 variant of the T4 helper lymphocyte and remains within infected cells throughout their life, being transmitted to other cells mainly by cell-to-cell contact. Other cells that may be affected include macrophages, and possibly endothelial, neuroglial, epithelial and dendritic cells. The principal effect of HIV infection on the immune system is depletion of CD4 lymphocytes (helper cells), which results in a drop in the absolute CD4 count and a reversal of the CD4/CD8 ratio. These are immune indicators of disease progression.
Oral manifestations (Figure 12.6)
Oral lesions are often early warning signs of HIV infection. Common disorders may manifest in different ways in the presence of HIV. In children, the most common lesions are:
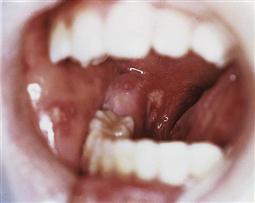
Candidosis
The most common oral lesion in HIV infection is acute pseudomembranous candidiasis. It is an early lesion and suggests the presence of other opportunistic infections. The severity of the candida infection may be related to the T4/T8 ratio and occurs when CD4 counts are <300/mL. Oesophageal candidiasis occurs when CD4 counts drop below 100/mL. Fungal infections can be related to reduced salivary flow and S-IgA. It responds well to treatment with systemic antifungals and an improvement in oral hygiene.
Ulceration
Recurrent herpes simplex infections are frequent and are typically intra-oral and circum-oral. Other parts of the body may also be affected. Aphthous-type ulcers are persistent and very common in children. Treatment is palliative with adequate hydration, analgesia and the use of Diflamm-C mouth rinses.
Atypical gingivitis
HIV-related gingivitis manifests as red erythematous gingival tissues and can extend to the free gingival margin. There is often spontaneous gingival haemorrhage and petechiae within the gingival margin, either localized or generalized. Consideration must be given to a fungal component. Treatment involves improved tooth brushing and flossing and the use of daily 0.2% chlorhexidine gluconate (Curasept) mouthwashes and gels.
Salivary gland enlargement
Parotitis or HIV associated parotid gland disease (HIV-PGD) occurs more frequently in paediatric than in adult patients and is similar to the presentation of mumps. It may be unilateral or bilateral and results in xerostomia and pain. Reduced salivary flow may lead to pseudomembranous candidiasis and dental caries. There have been mixed results with the use of antibiotics and glucocorticosteroids in treating this condition. Artificial saliva substitutes or oral lubricants can alleviate the xerostomia.
Hairy leukoplakia
This is uncommon in children, with only a few reported child cases. It occurs predominantly on the lateral border of the tongue and occasionally on the buccal mucosa and the soft palate.
HIV-related periodontitis
HIV-related periodontitis presents with deep pain and spontaneous bleeding, inter-proximal necrosis and cratering, and intense erythema more severe than acute necrotizing ulcerative gingivitis (ANUG). HIV periodontitis appears more frequently in HIV-infected patients who have reduced T4/T8 ratios and symptomatic opportunistic infection. Organisms such as black-pigmented bacteroides and Gram-positive bacilli, which are similar to those found in adult periodontitis, have been identified in HIV periodontitis.
Kaposi’s sarcoma
Uncommon in children and adolescents. The lesion mainly affects the palate, and also the gingivae and the tongue. Treatment is by chemotherapy, radiotherapy or laser excision.
Outcomes
Primary colonization by commensal organisms rather than reactivation of opportunistic infections usually occurs (cytomegalovirus, retinitis and toxoplasmosis are rare). Bacterial infections are also rare, although Streptococcus pneumoniae and Haemophilus influenzae are common respiratory complications. Kaposi’s sarcoma is seen infrequently but lymphomas (especially with central nervous system (CNS) involvement) can occur. The progression of disease process can vary and in many instances, oral and physical symptoms do not often present for years after infection with the immunodeficiency virus. Lymphocytic interstitial pneumonitis is frequently the cause of death for children with AIDS, but is often asymptomatic. There have been major advances in the management of HIV/AIDS with antiretroviral medications and consequently, many children may lead a normal and effective life.
Oncology
Childhood cancer accounts for about 1% of all cancer cases in the population. In Australia, the annual incidence of malignant tumours in children under 15 years is approximately 11 per 100 000 children. Approximately 600–700 children between birth and 15 years of age develop cancer each year. Whereas most adult cancers are carcinomas with strong aetiological associations, childhood cancers are a wide range of different histological types of tumour with less aetiological connection.
The incidence, either in childhood cancer as a whole or in individual types of cancer, varies little from one country to the next and no racial group is exempt. Among more than 50 types of childhood cancers, the most common forms include leukaemias, lymph/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


