12
Immunological and Mucocutaneous Disease
I. Background
The immune system helps protect the body against foreign agents. In the case of autoimmune diseases, the immune system attacks healthy cells in the body. The conditions discussed in this chapter are considered to be of immune origin, although other etiological agents may be identified in the future. The interactions of the immune system contribute to the magnitude of the disease process. When the immune system is triggered, a cascade of humoral and cellular immune responses is initiated. In many of these diseases, antibodies against normal epithelium are found in the circulation.
Description of Diseases/Conditions and Management
Allergy and Allergic Reactions
Allergic reactions are immune responses mediated by immunoglobulin E (IgE). These inflammatory reactions occur after repeated contact with an external antigen (allergen) in previously sensitized individuals. Some patients are more prone to severe and recurring reactions than others.
Contact Stomatitis
Hypersensitivity to allergens is common within the oral cavity. Allergens creating reaction of the oral mucosa include:
- foods;
- dental materials, flavoring, and other chemicals in toothpaste and mouthwashes;
- medications.
Clinical Features of Contact Stomatitis
- Oral mucosal erythema and edema.
- Gingiva with generalized uniform redness.
- Buccal mucosa might become puffy and dark red.
- Lips appear swollen, erythematous, and subject to chronic ulcerations.
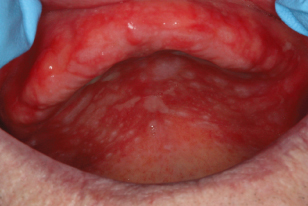
The patient usually complains of a burning sensation and sensitivity or irritation to hot, cold, alcohol, and spicy food. An allergy to denture base material occurs when the acrylic has been incompletely cured. See Fig. 12.1.
Figure 12.1 Contact stomatitis of the maxilla due to a denture allergy.
Angioedema
Angioedema is a clinical presentation of a group of allergic conditions with different etiological pathways. Angioedema usually develops as a regional, painless swelling of the lips, cheek, or tongue. It is of a great concern when the posterior anatomical structures are involved, because the airway becomes subject to compromise. Upon cessation of contact with the allergen, the swelling subsides usually within 24–48 hours. The two forms of angioedema are acquired and hereditary:
- Acquired angioedema is the most common form and is usually the result of a recent ingestion of a medication such as penicillin, nonsteroidal anti-inflammatory drugs (NSAIDs), or angiotensin-converting enzyme (ACE) inhibitors (e.g., lisinopril).
- Hereditary angioedema is a rare form of the disease, inherited as an autosomal dominant trait. In these patients, the swelling develops after mild trauma to the area.
Orofacial Granulomatosis (OFG)
OFG is an uncommon immunologically mediated disorder characterized by persistent or recurrent soft tissue enlargement, ulceration, and a variety of other orofacial features. The chronic inflammation of OFG is often characterized by the presence of granulomas in the subepithelial tissues. Risk factors include genetics, food allergy, dental material allergy, microbial etiology, and immunological etiology.
Graft-versus-Host Disease (GVHD)
GVHD occurs mainly in recipients of allogeneic hematopoietic stem cell transplantation (HSCT) for treatment of hematological diseases. See Chapter 8. Antigenic differences between the donor and the host will lead to graft rejection unless the donor cells in the graft are given advantage over host cells. Such advantage is achieved through suppression of the host immune cells by chemotherapy with or without irradiation in order to deplete the host immune system and allow successful engraftment of donor stem cells.
Clinical Features of GVHD
Both acute (aGVHD) and chronic (cGVHD) phases of this complication develop often involving multiple organs. The phases occur after different times following transplant; however, consensus criteria define each by their clinical characteristic and pathological features rather than chronologically1:
- aGVHD occurs within 100 days of HSCT, but can persist beyond that time or recur. It has a relatively uniform clinical picture, classically manifested by erythematous rash, diarrhea, and/or liver involvement; generally occurs early posttransplant; and is the major cause of early mortality.
- cGVHD typically occurs beyond day 100 posttransplant and can affect almost every major organ but most commonly involves skin, oral, vaginal, and conjunctival mucosa; salivary and lacrimal glands; and the liver. Approximately 40–70% of engrafted patients surviving the initial transplant will eventually develop cGVHD, which can persist for long periods of time and require long-term management from multiple disciplines.2
Risk Factors
- Prior diagnosis of aGVHD is the risk factor associated most consistently with subsequent cGVHD.
- Increasing donor and recipient age.
- Increasing (T cell) dose in the graft.
- Female donor and male recipient combination.
- Unrelated donors.
- Total body irradiation.
Clinical Similarity to Autoimmunity
In many cases, the manifestations of aGVHD and cGVHD resemble both clinically and histologically autoimmune disorders such as lichen planus (LP), Sjögren’s syndrome, and systemic lupus erythematosus1,3:
- Oral Findings in aGVHD
Mucosal erythema, ulcerations, and painful desquamative oral lesions occur often in patients undergoing immunosuppression for HSCT. However, a true clinical case definition of oral acute GVHD is lacking as several factors contribute to oral lesion development during the first few weeks following transplant.
- Oral Findings in cGVHD
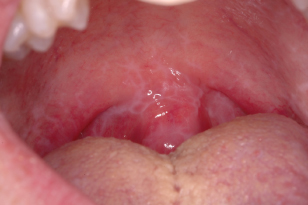
Classic features of oral cGVHD include lichenoid changes (see Fig. 12.2), ulcerations, salivary gland dysfunction, restricted oral opening, mucoceles, and rarely squamous cell carcinoma.
Figure 12.2 Reticular pattern of the soft palate and uvula associated with chronic graft-versus-host disease (GVHD) in a child post-bone marrow transplant.
Treatment of GVHD
Multiple factors must be considered when treating a patient with oral GVHD. The clinician may need to try various treatments and possibly treatment combinations to manage oral GVHD symptoms. Pharmacotherapy for oral GVHD may be systemic, topical, or injectable. The two systemic immunosuppressive drugs used most commonly are cyclosporine and corticosteroids, either alone or in combination. Other systemic agents are also used.
Topical corticosteroids such as clobetasol or fluocinonide may be used for local management for oral cGVHD.
Vesiculobullous Conditions
Vesiculobullous diseases are a group of severe, potentially life-threatening diseases, characterized by blisters and erosions of skin and/or mucous membranes. In these conditions, autoantibodies are formed that will impact different components of the mucous membrane or skin. Based on histopathological, immunological, and clinical criteria, autoimmune bullous diseases are classified into two major groups associated with autoantibodies to desmosomal (pemphigus group) or hemidesmosomal proteins (subepidermal blistering diseases, e.g., pemphigoid diseases and epidermolysis bullosa acquisita [EBA]).
Pemphigus Group and Pemphigus Vulgaris (PV)
Pemphigus is a group of rare, potentially life-threatening autoimmune mucocutaneous diseases that are characterized by blistering that affects stratified squamous epithelium and results in cutaneous or mucosal blistering, or both. It affects less than 0.5 patients/100,000 population/year,4 and there are several variants. PV is the main variant and the one that usually affects the mouth. The remaining of the discussion relates to PV.
Etiology and Pathogenesis of PV
- Most cases are idiopathic; isolated cases have an identifiable trigger such as diet or drugs (ACE inhibitors, NSAIDs, and some antibiotics).
- A significant number of cases show a strong genetic, as well as ethnic relationship, primarily within the Ashkenazi Jews and those of the Mediterranean descent.5
- The pathological process is mediated by autoantibodies that target the extracellular adhesion components, which in case of oral PV is mainly desmoglein 3.6
Clinical Features of PV
- The oral mucosa is usually affected at an early stage in PV.
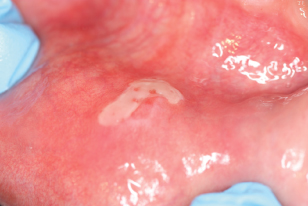
- Blisters, which eventually lead to chronic erosions and ulcers, are seen mainly on the buccal mucosa, palate, ventral surface of the tongue, and lips. See Fig. 12.3.
- Advanced stages consist of severe desquamative or erosive gingivitis.
- Oral lesions are almost invariably followed by lesions on the skin.
- PV may be occasionally associated with other autoimmune disorders, particularly rheumatoid arthritis, lupus erythematosus, or Sjögren’s syndrome.
Figure 12.3 Area of ulceration on the lower labial mucosa seen in a patient with pemphigus vulgaris.
Diagnosis of PV
- In patients with PV and active blistering, firm sliding pressure separates normal-looking epithelium (Nikolsky sign), but this is neither sensitive nor specific.
- Biopsy of the perilesional tissue with histological examination and immunostaining is crucial.
- Assay of serum antibody titers by indirect immunofluorescence may also help guide prognosis and treatment.
Treatment of PV
Treatment is initially aimed at bringing the disease under control rapidly until it is possible to reduce it gradually. Treatment is invariably with systemic corticosteroids; other treatments include cyclosporine, dapsone, tacrolimus, rituximab, and intravenous immunoglobulins in steroid-resistant PV.
Mucous Membrane Pemphigoid (MMP)
MMP is a chronic autoimmune subepithelial blistering disease. MMP can be localized or extensive and can affect both mucosal and cutaneous surfaces.7
Epidemiology of MMP
- Found in 2–5 per 100,000 population a year.
- Occurs twice as often in women as men.
- Primarily affects middle-aged and older adults. However, children may also be affected.
Pathogenesis of MMP
In MMP, autoantibodies attack antigen sites in the molecules connecting the epithelium to the connective tissue and prevent the linkage of molecules in the hemidesmosomes. The major antigens involved in oral MMP are believed to be BP180 and laminin 5.
Clinical Features of MMP
- MMP may arise at any mucosal site, most commonly the oral and conjunctival mucosa.
- Eighty-five percent of cases will have oral involvement without concomitant skin involvement, and it may be the only site of disease.
- Lesions usually involve the gingival, palatal, and buccal mucosa, and less over the tongue and lips.
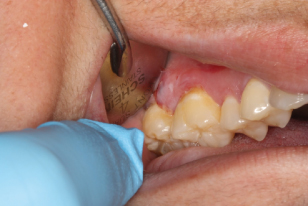
- The gingival presentation is typically painful erythematous and tender erosions with desquamation, either spontaneously or with very minimal physical trauma, such as with toothbrushing. Often, there is an inability to maintain oral care with consequent heavy accumulation of plaque and an additional inflammatory burden from this source. Small vesicles may be observed that rupture easily, but in comparison to those of PV, are long lasting and well defined. See Fig. 12.4.
- Over time, there may be scarring at the sites of repeated vesiculobullous lesion development and healing, mainly over the posterior soft palate.
Figure 12.4 Erosive gingival lesion in a patient with mucous membrane pemphigoid.
Treatment of MMP
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


