11
Skin disorders
Several skin diseases can involve the mouth or the head and neck, and can be seen by dentists during routine dental examination. Dentists should encourage a patient to see his physician, dermatologist or an oral medicine specialist for further evaluation and possible treatment if an abnormality is noted. Some diseases can be monitored and managed directly by a general dental practitioner.
In this overview, diseases are classified as infections, immunologically mediated eczematous lesions, facial eruptions, pigmented lesions (including melanoma) and malignant epidermal lesions.
Infections
Bacterial infections
Impetigo
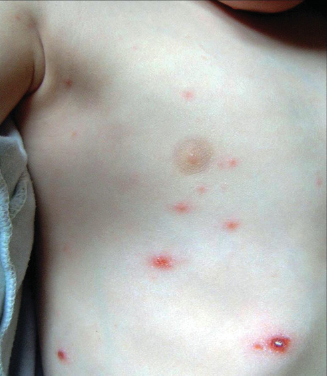
Impetigo is a superficial infection of the skin by Staphylococcus aureus or Streptococcus pyogenes, or both. It is the most frequent bacterial dermatitis in infancy, occurring mainly in pre-school-aged children, particularly during the summer. Impetigo is very contagious and it is not unusual to observe minor epidemics in nurseries. Transmission is mainly by direct patient to patient contact and is favoured by environmental factors (such as high temperature and humidity, overcrowding or poor hygiene) and personal factors (such as malnutrition, poor hygiene, debilitating diseases or systemic therapies with antibiotics and immunosuppressants). Moroever, impetigo often develops in pre-existing skin diseases as chickenpox (Fig. 11.1), insect bites, abrasions, ulceration, burns, eczema and pediculosis. In these cases it is more appropriately termed a secondary impetigo.
Impetigo is classified as non-bullous or bullous. Non-bullous impetigo predominates and presents with an erosion (sore), cluster of erosions, or small vesicles or pustules that have an adherent or oozing honey-yellow crust. This form may be caused by staphylococci or streptococci. Bullous impetigo presents as a large thin-walled bullae (2–5 cm) containing serous yellow fluid. These often rupture, leaving a complete or partially denuded area with a ring or arc of remaining bulla. Bullous impetigo is almost always caused by S. aureus.
Impetigo is commonly seen on the face, especially below the nostrils, but it may occur in any area. Topical muporicin may be given for localised disease, whereas for widespread involvement an oral antibiotic (cephalexin, dicloxacillin or erythromycin) is effective.
Syphilis
Syphilis is a sexually transmitted disease caused by Treponema pallidum. The incidence of syphilis has declined dramatically since the introduction of penicillin. In the last 20 years, however, there has been a resurgence largely due to concurrent HIV infection.
Syphilis is characterised by several stages – primary, secondary and tertiary. They are associated with oral lesions, but the lesions vary with the stage. The principal route of syphilis infection is through sexual contact, although it can be spread transplacentally, leading to foetal death, premature delivery or congenital syphilis.
Primary syphilis is associated with a lesion known as the ‘chancre’. This occurs at the site of penetration of the bacterium into the mucosa after 3–4 weeks. It is usually localised on the genitalia, but in about 10% of the cases the chancre occurs extragentially, mainly in the oral mucosa. The most frequent oral site of chancre is the lips, followed by the tongue and more rarely by the palate and tonsillar areas. It begins as a solitary firm nodule that soon erodes, leaving a painless ulcer with raised border and indurated base. Regional lymph nodes are enlarged, normally unilaterally, and they are mobile, hard and not tender. If not treated, the chancre heals spontaneously without scarring in 3–8 weeks.
Figure 11.1 A child with chickenpox. The crusted lesion in the lower right part of the picture is an old lesion. When all the lesions have crusted over, they are no longer infectious.
Secondary syphilis develops 1–4 months after the infection. The oral lesions are diverse and may be nonspecific. These include malaise, low-grade fever, a pharyngitis, loss of appetite, weight loss, polyarthralgias and myalgias, and a generalised lymphadenopathy with splenomegaly. Mucocutaeneous manifestations include a variable asymptomatic skin rash, symmetrical and generalised, consisting of several small erythematous macules and papules on the face, trunk, palms, soles and genital regions. It may last for hours or weeks and disappear without scarring.
Mucous patches are the most frequent oral manifestations of secondary syphilis. These comprise shallow, irregular, sometimes ulcerated plaques covered by a grey-white necrotic membrane with surrounding erythema. Lesions may be occasionally painful. Snail-track ulcers result when multiple mucous patches become confluent.
Lesions of tertiary syphilis develop after a period of 4–7 years or more. The onset is insidious, and during the latent period the patient may appear well. The characteristic lesion of tertiary syphilis is a local destructive granuloma called a ‘gumma’ or a glossitis with mucosal atrophy, which may undergo malignant transformation.
Gummas appear initially as painless swellings which undergo necrosis leaving indolent deep ulcers. Sites of predilection are the legs, scalp, face and chest. In the oral cavity, gummas are frequently located on the palate, perforating the hard palate.
Congenital syphilis is transmitted by the mother to the foetus in utero. Clinical signs are corneal ulcers and opacities, high-arched palate, short mandible, commissural rhagades, saddle nose, pegged lateral and notched central incisors (Hutchinson’s teeth) and molar teeth with alternating non-anatomical depressions and rounded rudimentary enamel cusps on the crown surface (mulberry or Moon’s molars).
Patients suspected of having syphilis are initially tested using a non-treponemal antigen test. If this is positive, confirmation using a treponemal-specific test such as the T. pallidum haemoagglutination assay (THPA) is done.
Antibiotics, mainly parenteral penicillin G, are the mainstay of treatment. Non-pregnant, penicillin-allergic patients who have primary or secondary syphilis may use doxycycline and tetracycline. No proven alternatives to penicillin are available for treating neurosyphilis, congenital syphilis or syphilis during pregnancy. Penicillin is also recommended in HIV-infected patients.
Viral infections
Molluscum contagiosum
Poxvirus, responsible for molluscum contagiosum (MC), is a DNA virus that replicates in the cytoplasm of infected cells. MC is a benign lesion usually seen on children’s skin. Males are affected more frequently than females. Swimming in public pools is correlated with incidence. MC is most commonly found in immunocompromised patients. The lesions are grouped, minute, dome-shaped, pink to flesh-coloured, umbilicated 3–5 mm papules mainly occurring in adults on the face, trunk and genitalia. In children, they may occur anywhere. Very rarely MC may affect the oral cavity on the lips, buccal mucosa and palate.
Histopathological examination establishes the final diagnosis. Surgical excision with cryotherapy is the preferred mode of treatment of MC in adults. In children treatment is not always needed as the normal course of MC is spontaneous resolution.
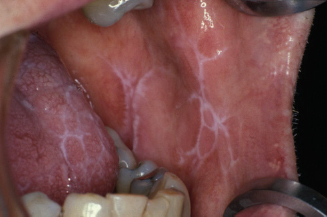
Figure 11.2 Oral lichen planus.
Immunologically mediated skin diseases
T cell-mediated diseases
Lichen planus
Lichen planus (LP) is an inflammatory mucocutaneous condition which most commonly affects middle-aged adults of both sexes, with a slight predominance in women. The prevalence of lichen planus is unknown, but it is estimated to occur in <1% of the population. It is thought to be significantly less frequent than exclusive oral LP (OLP) that affects approximately 1–2% of the population (Fig. 11.2). Estimates of prevalence vary among different populations, but the condition does not appear to exhibit a racial predilection.
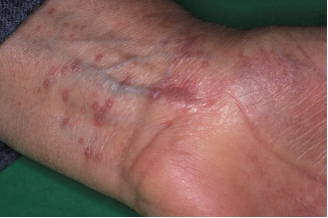
The classic appearance of skin lesions includes violaceous polygonal flat-topped papules and plaques frequently involving the flexor surfaces of the extremities, particularly the wrists. Close examination reveals a reticulated pattern of white scale known as Wickham’s striae (Fig. 11.3). The lesions may mimic discoid lupus, psoriasis or secondary syphilis. Another clue to the diagnosis is that lesions may occur in areas exposed to trauma (Koebner’s phenomenon). In most patients the lesions cause intense pruritus (itching), a hallmark of LP.
While LP often occurs only on cutaneous surfaces, it may also involve the oral mucosa, the genital mucosa, the nails and the scalp, and less commonly oesophageal and conjunctival mucosae. Moreover, these areas may be exclusively involved.
Multiple types of nail changes may occur that include ridging, distal splitting, thinning, subungual hyperkeratosis and even loss of the nail. None of these findings are specific to LP.
Figure 11.3 Skin lesions in a patient with lichen planus.
Lichen planopilaris represents LP involvement of the scalp and hair follicles. Patients usually present with the initial clinical sign of areas of hair loss with keratotic follicular papules. If left untreated, these areas progress to scarring alopecia. The hair follicles are destroyed and regrowth of hair will not occur, resulting in scarring alopecia.
OLP lesions usually have recognisable and distinctive features and a characteristic distribution. OLP may manifest in one of the following clinical forms: papular, reticular, erythematous (atrophic) and erosive (ulcerated, bullous). Whereas papular/reticular lesions occur as isolated lesions and are often the only clinical manifestation of the disease, erythematous lesions are accompanied by reticular lesions, and erosive lesions are accompanied by reticular and erythematous lesions in most of the cases. This feature helps clinically to differentiate OLP from other erosive diseases such as pemphigus and pemphigoid, which are characterised by isolated areas of erythema and/or erosions.
The reticular lesions appear as a network of connecting and overlapping white lines (so-called Wickham’s striae), papules or plaques. Although some patients may display an impressive array of diffuse and widespread reticulated lesions, they rarely complain of symptoms and often are unaware of their presence.
Erythematous and erosive OLP lesions result in varying degrees of discomfort. The number of ulcerations is variable, as are their size and location. The erosive lesions hardly ever remit spontaneously and may lead to confusion with other autoimmune diseases such as pemphigus and pemphigoid, which share similar clinical features. The posterior buccal mucosa is the most frequent site of involvement, followed by the tongue, gingiva, labial mucosa and vermilion of the lower lip. Lesions on the palate, floor of the mouth and upper lip are uncommon. Approximately 10% of patients with OLP have the disease confined to the gingiva frequently resulting in the so-called desquamative gingivitis (DG) appearance because of the erythematous and erosive features. Lesions resembling those observed in other diseases including pemphigoid, pemphigus and foreign body gingivitis are not easily identified as OLP unless there are co-existent reticular lesions on the gingiva or elsewhere in the oral cavity.
The most frequent extraoral site of involvement in female patients with LP is the genital mucosa. The association of LP of the vulva, vagina and gingiva is recognisd as the vulvovaginal–gingival syndrome. When LP affects the genital mucosa, erosive lesions are predominant, although asymptomatic reticular lesions can be identified in a quarter of all patients.
The aetiology of LP is unknown, although many studies have investigated and support an immunological pathogenesis. Current data suggest that LP is a T cell-mediated disease. While most cases of LP are idiopathic, some are linked to medication use (mainly antimalarials, antidiabetic agents, β-blockers and NSAIDs), HCV infection or dental restorative materials (mainly amalgam).
Lichenoid lesions (mainly involving oral and genital mucosae) have a small but significant pre-malignant potential, probably <1%. The increased risk of cancer appears to be independent of the clinical type of LP and therapy administered.
A diagnosis of LP is reached on the basis of the typical clinical appearance, and a biopsy is recommended to confirm the clinical diagnosis in doubtful cases and mainly to exclude dysplasia and malignancy in oral/genital mucosae. Gingival LP may be more difficult to diagnose, and direct immunofluorescence of perilesional mucosa may facilitate the diagnosis and exclude other causes such as bullous diseases.
While the natural history of cutaneous LP is often one of remission within a year, OLP rarely undergoes spontaneous and permanent disappearance. Active treatment is indicated for symptomatic LP. Potent topical corticosteroids are used to treat the early or localised lesions of patients with both cutaneous and mucosal LP, whereas oral corticosteroids are the most common treatment for patients with generalised cutaneous or mucocutaneous LP, or for recalcitrant disease, although recommendations concerning dosage and duration of therapy vary.
Psoriasis
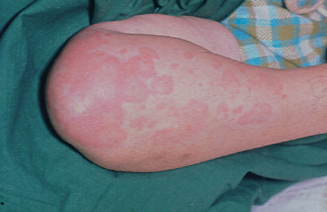
Psoriasis is an immune-mediated, papulosquamous skin disorder. Approximately 1–3% of the world population is affected. The severity varies from a minor, localised and intermittent skin eruption (Fig. 11.4) to total skin involvement and associated systemic effects.
Figure 11.4 A patient with psoriasis.
A substantial proportion of patients with psoriasis will develop a form of inflammatory arthritis known as psoriatic arthritis, which contributes significantly to the patient’s disease burden, with physical disability, pain and reduction in quality of life. Psoriasis can present at any age, although it is thought to exhibit a bimodal distribution of age of onset. One occurs at an early age (late teens to twenties), having frequent relapses as well as more extensive body involvement and more frequent nail involvement. The later group (sixth– seventh decades) displays a more stable clinical course. In the younger group a familial tendency is common. Psoriasis occurs with increased frequency in association with Reiter’s syndrome, inflammatory bowel disease, pemphigus and pemphigoid.
The aetiology of psoriasis is still unclear, but several drugs (lithium antimalarials and β-blockers) and streptococcal infection may trigger the disease. Similarly to LP, minor skin injury such as scratches or burns may trigger psoriasis (Koebner phenomenon). Psoriasis is considered a T cell-mediated disorder, and some increased inflammatory mediator produced by those cells, such as interleukin-8, can explain some characteristic features of psoriasis, e.g. the influx of neutrophils (causing small microabscesses or pustules within the epithelium) and the enhanced epidermal proliferation.
Clinically, psoriasis is characterised by thick silvery scales covering an erythematous plaque and typically involving some body sites, notably elbows, knees, scalp and sacrum. The face is uncommonly affected by psoriasis, except in severe cases.
The different treatment options for psoriasis are vast. Which therapy is best for a patient depends a great deal on the severity, location, accessibility of treatment and patient preference. For milder disease, there are topical therapies, mainly using corticosteroids, vitamin D3 analogues, retinoids (vitamin A derivatives), salicylic acid, calcineurin inhibitors (ciclosporin and tacrolimus) and dithranol. For more moderate to severe disease, photo (chemo) therapy and systemic treatments such as methotrexate and ciclosporin are used.
Connective tissue diseases
Lupus erythematosus
The term lupus erythematosus (LE) refers to a connective tissue disease comprising three subsets:
All can give rise to oral lesions, which may appear similar to those of OLP.
Systemic lupus erythematosus (SLE)
SLE is a multisystem disease of unknown cause that is characterised by the presence of multiple autoantibodies, mainly antinuclear antibodies. Typically, it affects the skin, joints, kidneys, lungs, nervous system, serous membranes and other systems. The clinical course is extremely variable, and ranges from life-threatening to mild disease. The disease is predominantly present in women, especially those in their twenties and thirties, but it may occur in children and the elderly.
Cutaneous manifestations of SLE are present in 85% of patients during the course of the disease. The typical localised skin lesion of LE is erythema over the malar eminences of the face and bridge of the nose (the so-called butterfly rash). The nasolabial folds are typically spared. The butterfly rash has been noted in 52% of patients with SLE at the time of diagnosis. It is frequently associated with exposure to sunlight or artificial sources of UV light. The lesions typically heal well without scarring. Less frequently, SLE may give rise to a diffuse or papular erythema of the face, upper trunk or extremities that resembles a viral rash or drug eruption. Lesions develop quickly and last for hours to days. The development of this rash is most often associated with systemic disease activity and is usually related to sun exposure. Facial involvement can be extensive.
Oral lesions appear in about 20–30% of cases of SLE and can, rarely, be the presenting sign. The typical oral lesions are characterised by central atrophic areas or shallow erosions, radiating white striae at margins characteristically far less sharply defined than in OLP. They are often patchy and unilateral, and may be on the hard palate, which OLP typically spares.
Subacute cutaneous lupus erythematosus (SCLE)
SCLE presents with erythematous macules and papules that develop into papulosquamous or annular plaques.
SCLE is highly photosensitive, with lesions being seen most commonly on the V area of the neck and upper chest, upper back, shoulders, extensor surfaces of the arms and forearms, and dorsum of the hands (the knuckles are typically spared). The face and scalp are much less commonly involved. Patients with SCLE often have mild systemic disease. This systemic activity is mainly musculoskeletal with associated serological evidence of disease, most commonly anti-Ro antibodies. The oral lesions are similar to those of SLE.
Discoid lupus erythematosus (DLE)
DLE occurs in 20% of patients with SLE. Most patients having DLE lesions (Fig. 11.5) never develop features of SLE. DLE lesions are most commonly on the scalp (eventually causing a scarring alopecia), face, ears, V region of the neck, and extensor surfaces of the arms. Nail involvement in DLE is also common. Oral mucosa is involved in 15–20% of cases. The buccal mucosa, the vermilion and the gingiva are usually affected. The oral lesions are similar to those seen in SLE but usually less severe. They can be the sole manifestation of the disease.
The occurrence of typical LP lesions associated with LE (overlapping syndrome) can occur.
LE is a multifactorial disease, and genetic, immunological, infectious, hormonal and other environmental factors may modulate the disease expression and clinical course.
Investigations in LE aim to achieve a diagnosis and to determine the presence and the degree of internal organ involvement. Skin or oral biopsy normally shows a rather lichenoid inflammatory infiltrate, but this is highly variable in density and typically extends deeply in the connective tissue and may have a perivascular distribution. The diagnosis should be confirmed by the pattern of autoantibodies. The most specific is that to double-stranded (native) DNA. Other haematological findings include raised ESR, anaemia and often leukopaenia or thrombocytopaenia. Tests for internal organ involvement may include urinalysis, serum electrolytes, liver function tests, and others as determined by symptoms.
Figure 11.5 A patient with DLE.
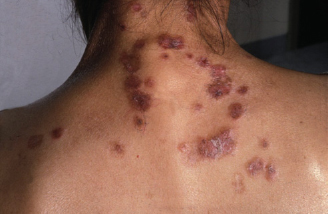
Oral lesions of DLE may respond to topical or systemic corticosteroids and topical calcineurin inhibitors. SLE may require other agents such as antimalarials and/or immunosuppressants.
Scleroderma
The term ‘scleroderma’ refers to a rare spectrum of diseases characterised by skin sclerosis. It encompasses both local and systemic sclerosis. Systemic sclerosis (SSc) is a multiorgan collagen vascular disease with a progressive, potentially fatal course. While the cause is unknown, microvascular changes, disturbed immunomodulation and an overproduction and deposition of collagen play central roles. Ssystemic sclerosis is relatively rare, with a reported incidence of 2–20 per million population. It mainly affects middle-aged women. The clinical manifestations of SSc include Raynaud’s phenomenon (cyanosis and blanching of the digits resulting from vasoconstriction) as well as fibrotic complications of the skin (Fig. 11.6), musculoskeletal, gastrointestinal, pulmonary, renal and cardiac systems.
The skin becomes thinned, stiff, pigmented and marked by telangiectasis. Skin thickening in systemic sclerosis begins in the fingers, but the head and neck region is involved in >70%. Fibrosis of the salivary and lacrimal glands leads to xerostomia and dry eyes. Subcutaneous collagen deposition in facial skin results in characteristic smooth, mask-like facies. Scarring of the eyelids causes a chronic widening of the palpebral fissures. The lips may be constricted (microstomia). Facial and mucosal fibrosis and involvement of the TMJ compromises oral access, making oral hygiene and dental care extremely difficult. The tongue can become stiff and narrowed, and periodontal ligament spaces may be widened. The mandibular angle may be resorbed, or rarely an extensive resorption of the jaws can be seen.
An intermediate form of systemic sclerosis is the CREST syndrome (calcinosis, Raynaud’s phenomenon, (o)esophageal involvement, sclerodactyly and telangiactasia).
Localised sclerosis is more common than systemic sclerosis. It affects females more commonly, with an average age at diagnosis of 33 years. Localised sclerosis may be linear and characteristically located on the midline of the forehead. Sclerotic patches of skin hardening with variable dyschromia occur, which may be localised or generalised, symmetrically involving the trunk and limbs. Involvement of visceral organs is uncommon in localised scleroderma, but Raynaud’s phenomenon is the initial presentation in about 70% of cases.
Whereas localised sclerosis is frequently diagnosed clinically, tests to confirm systemic sclerosis include skin biopsy, chest X-ray, renal function/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


