Diseases of Bone and Joints (Non-neoplastic and Non-infectious Disorders of Bone, Skeletal Dysplasias/Dysostoses, Constitutional Bone Disorders)
 . Group I: Defects in Extracellular Structural Proteins
. Group I: Defects in Extracellular Structural Proteins
 . Diseases of Bone of Questionable Etiology
. Diseases of Bone of Questionable Etiology
 . Diseases of Temporomandibular Joint
. Diseases of Temporomandibular Joint
 . Development Disturbances of Temporomandibular Joint
. Development Disturbances of Temporomandibular Joint
 . Traumatic Disturbances of Temporomandibular Joint
. Traumatic Disturbances of Temporomandibular Joint
 . Inflammatory Disturbances of Temporomandibular Joint
. Inflammatory Disturbances of Temporomandibular Joint
 . Neoplastic Disturbances of Temporomandibular Joint
. Neoplastic Disturbances of Temporomandibular Joint . Loose Joint Bodies
. Loose Joint Bodies . Temporomandibular Disorders
. Temporomandibular DisordersSkeletal dysplasias are a heterogeneous group of disorders, which result in disproportionate short stature. The nomenclature of these disorders remains confusing. In an attempt to develop uniformity, an international nomenclature and classification was proposed in 1969 and then updated many times later. In the 1992 revision, the classification was based on radiodiagnostic and morphologic criteria. In the 1997 revision, the groups of disorders were rearranged based on current etiopathogenetic information regarding the gene and/or protein defect in these disorders (Table 17-1). In the 2001 revision, the term dysostoses was incorporated in the nomenclature. All these revisions merely reflect the complexity of skeletal-genetic phenotypes. Over the recent years the accumulation of knowledge on genes and proteins responsible for genetic disorders of the skeleton has been unprecedented. A molecular pathogenetic classification of skeletal dysplasias based on the structure and function of the causative gene and protein was recently proposed (Table 17-2).
Table 17.1
International nosology and classification of genetic disorders of bone —2006
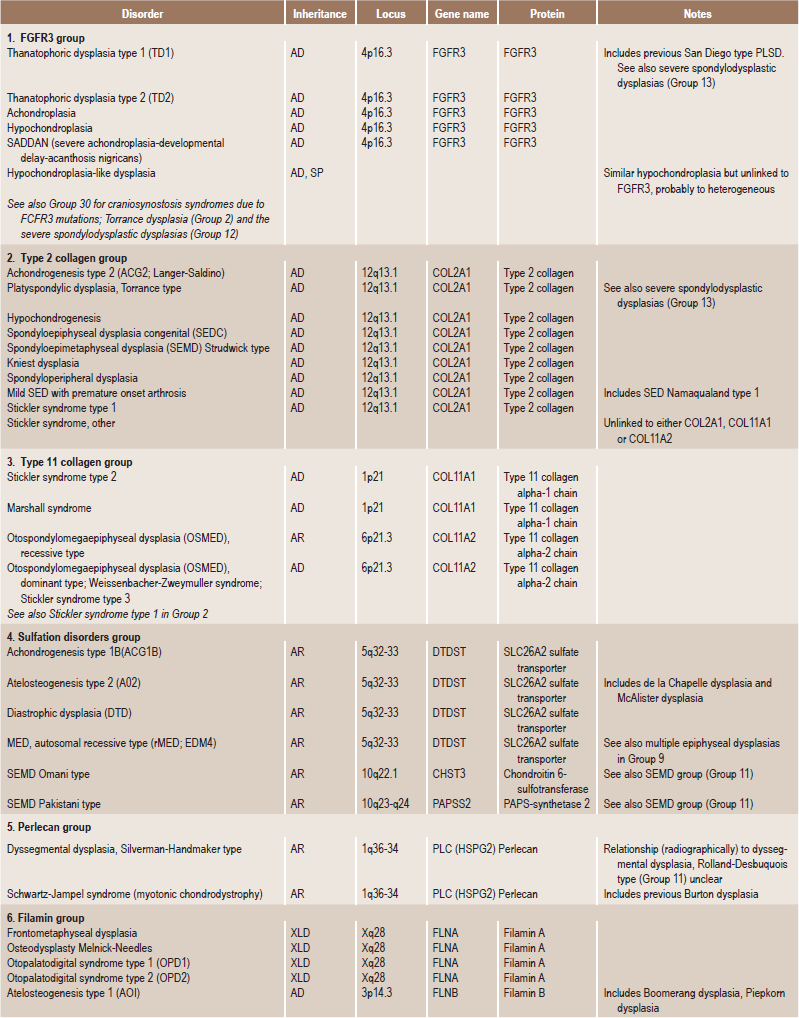
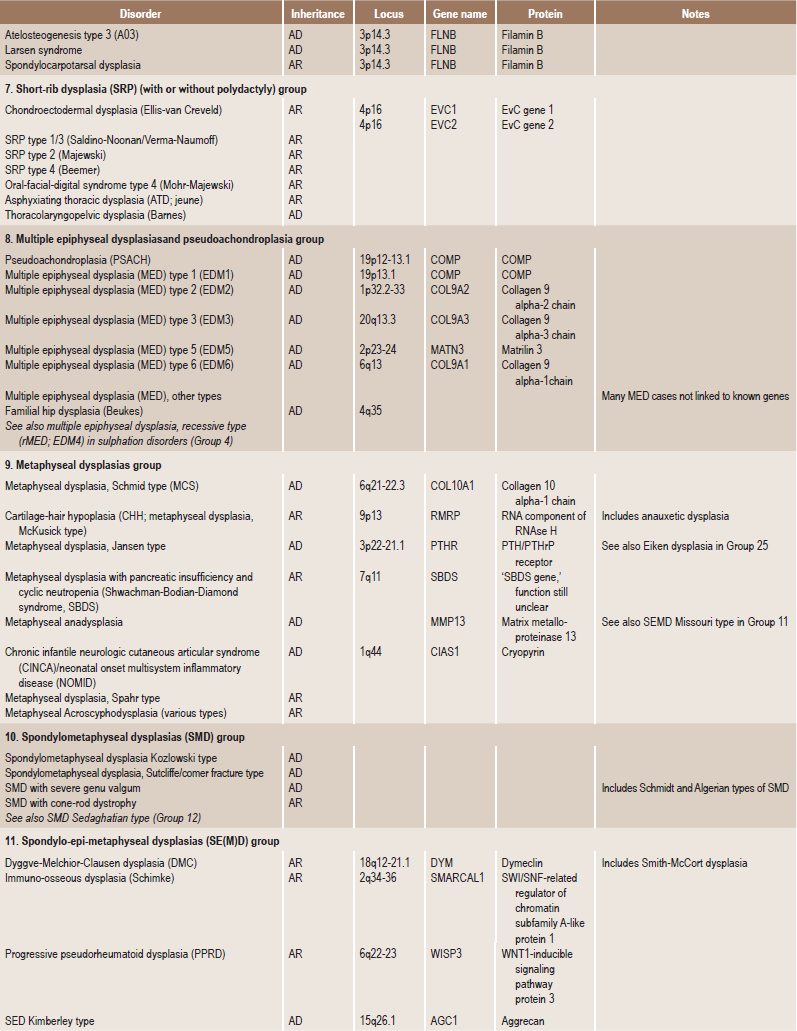
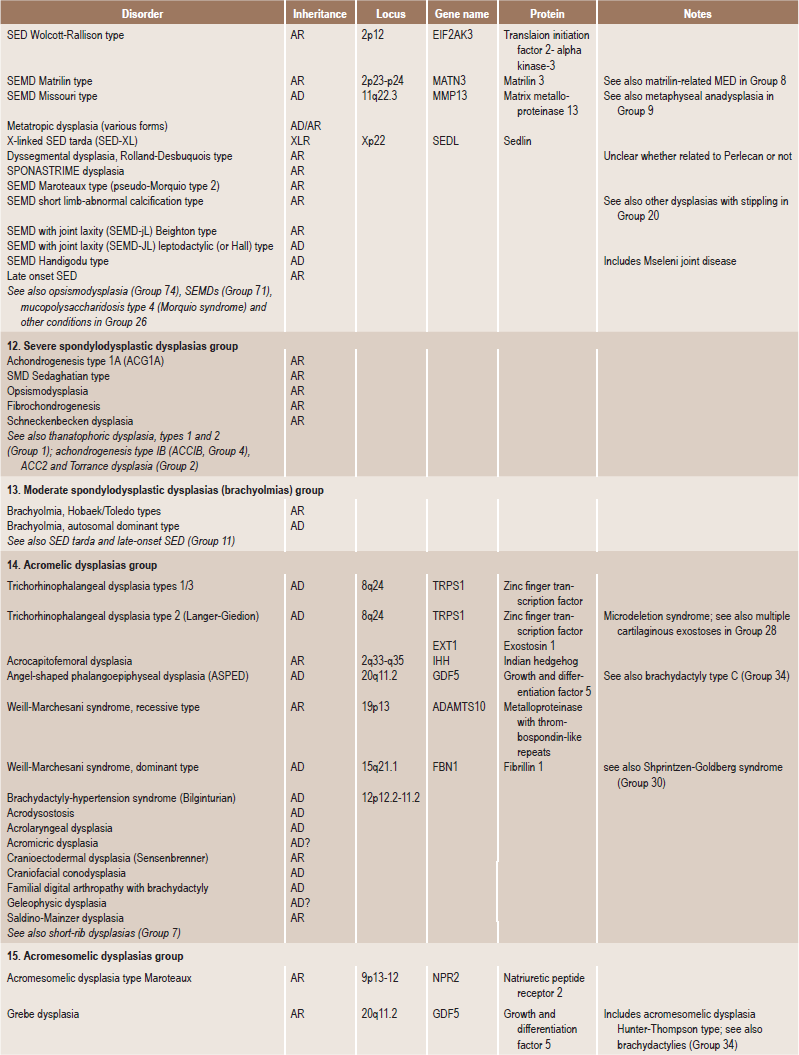
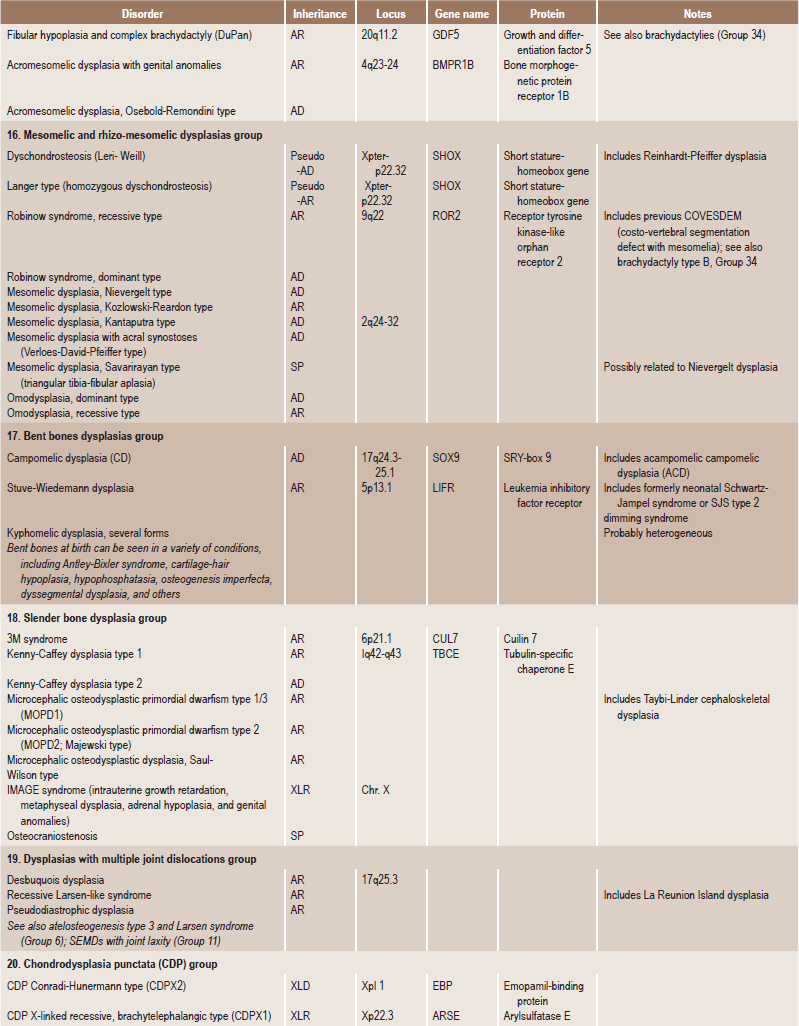
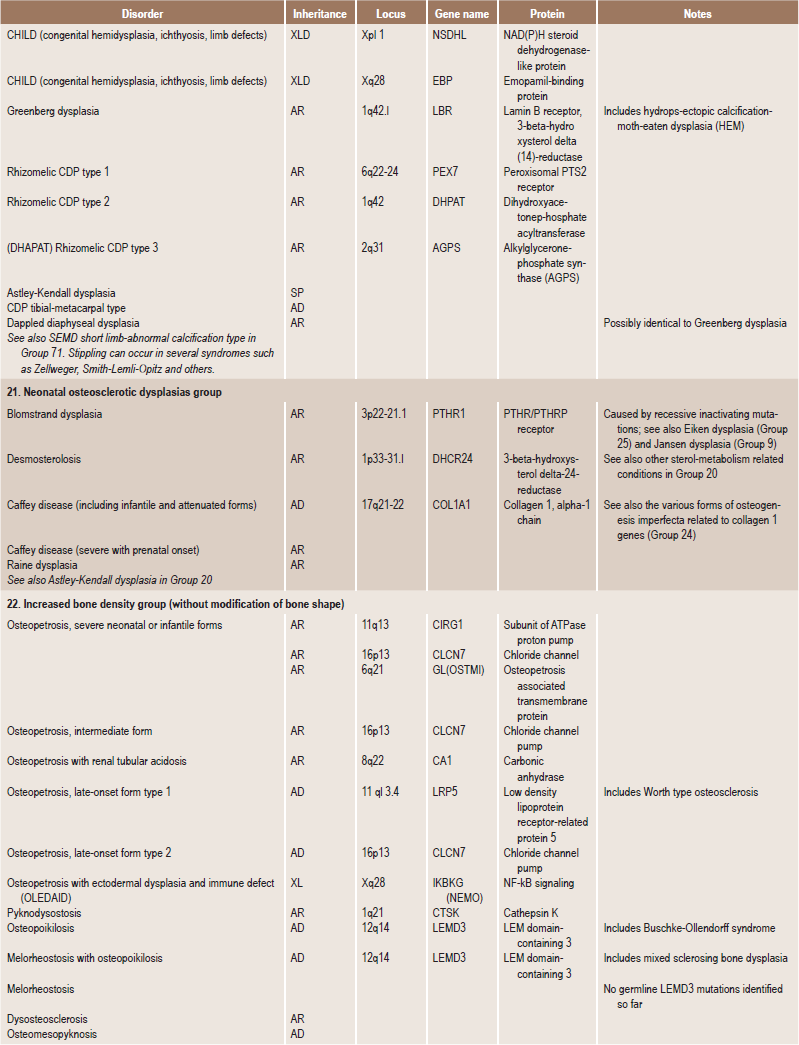
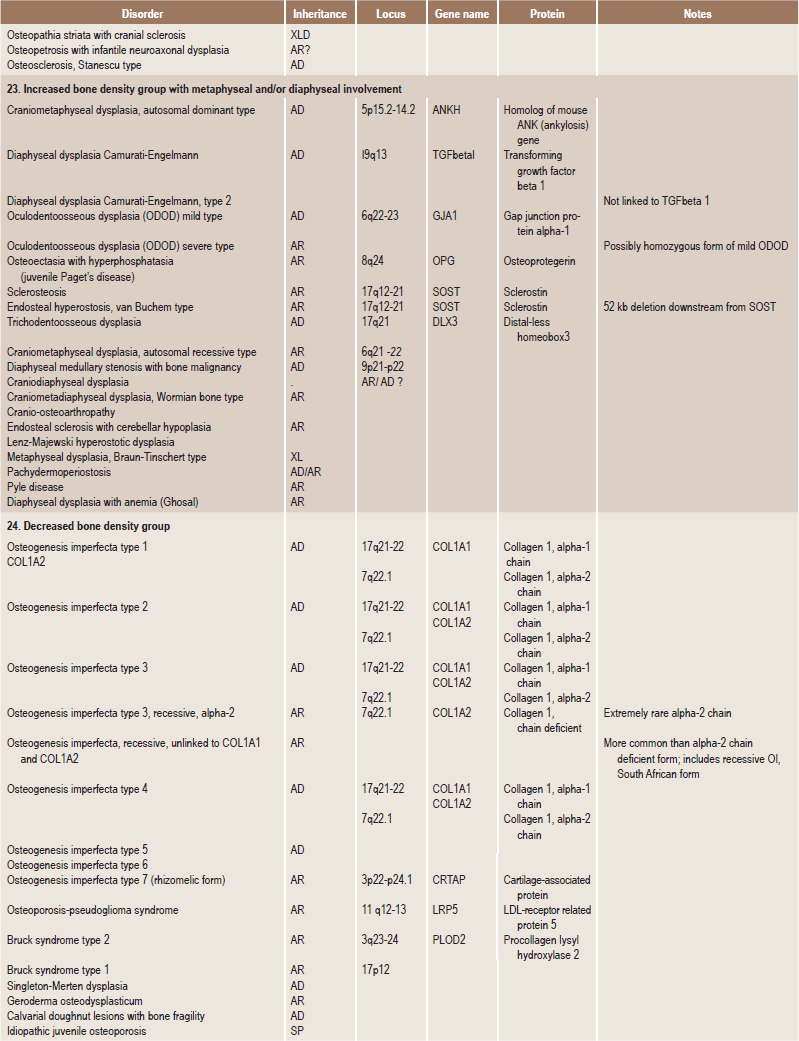
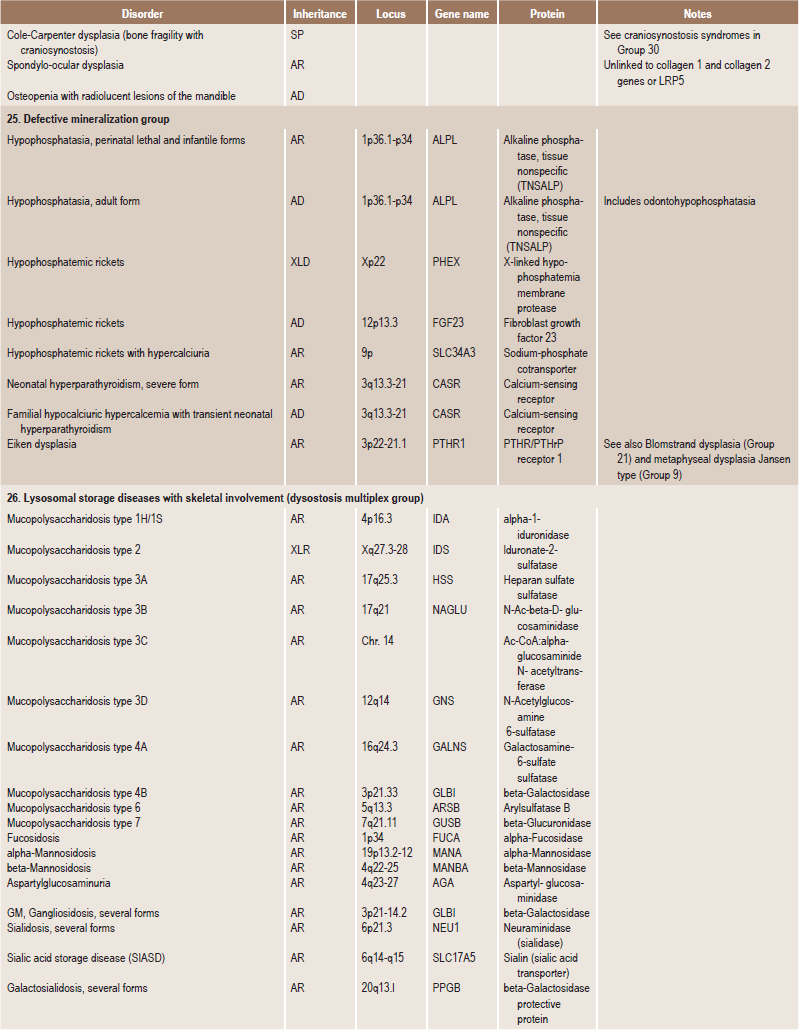
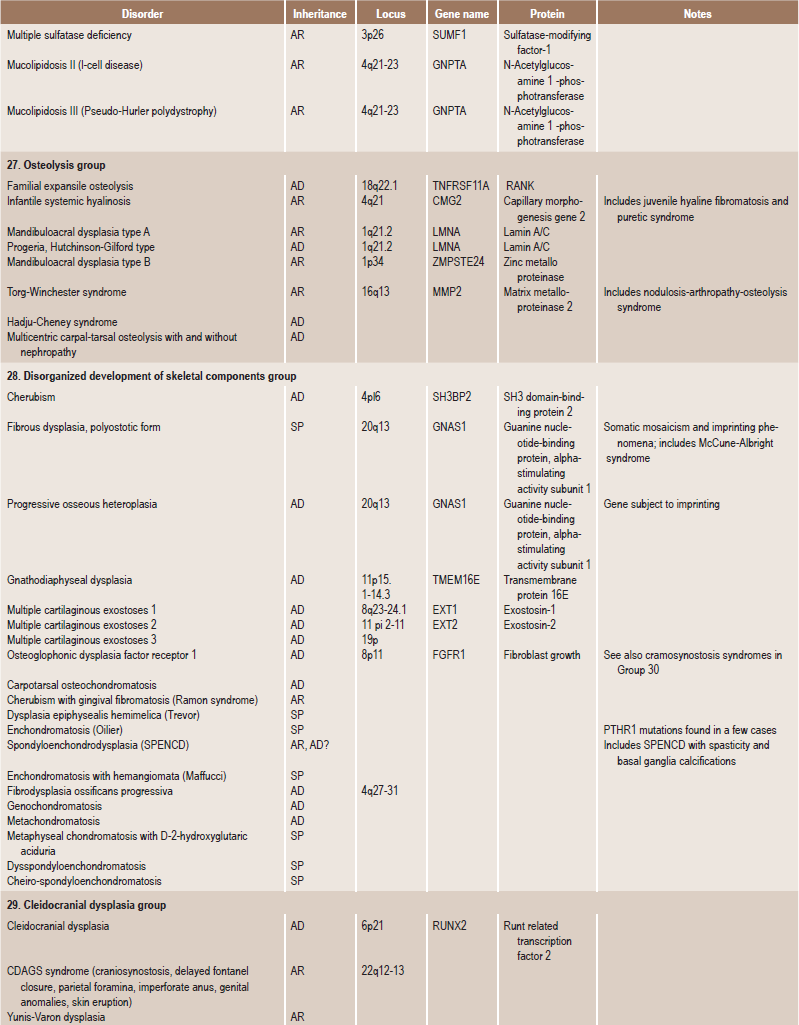
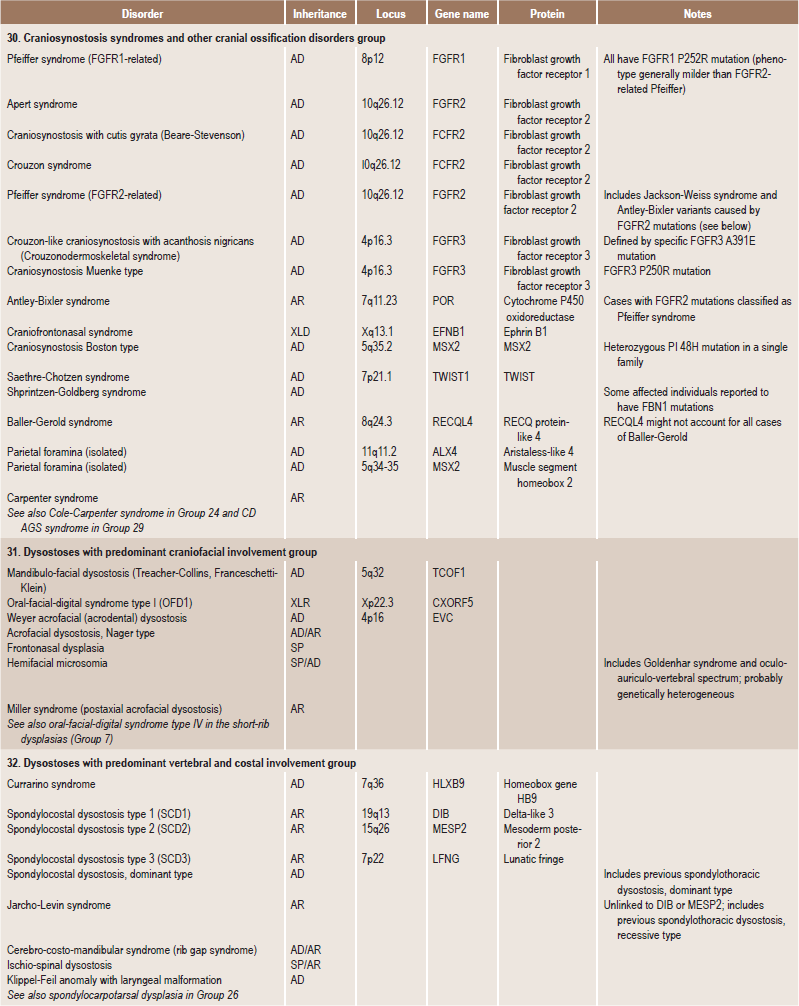
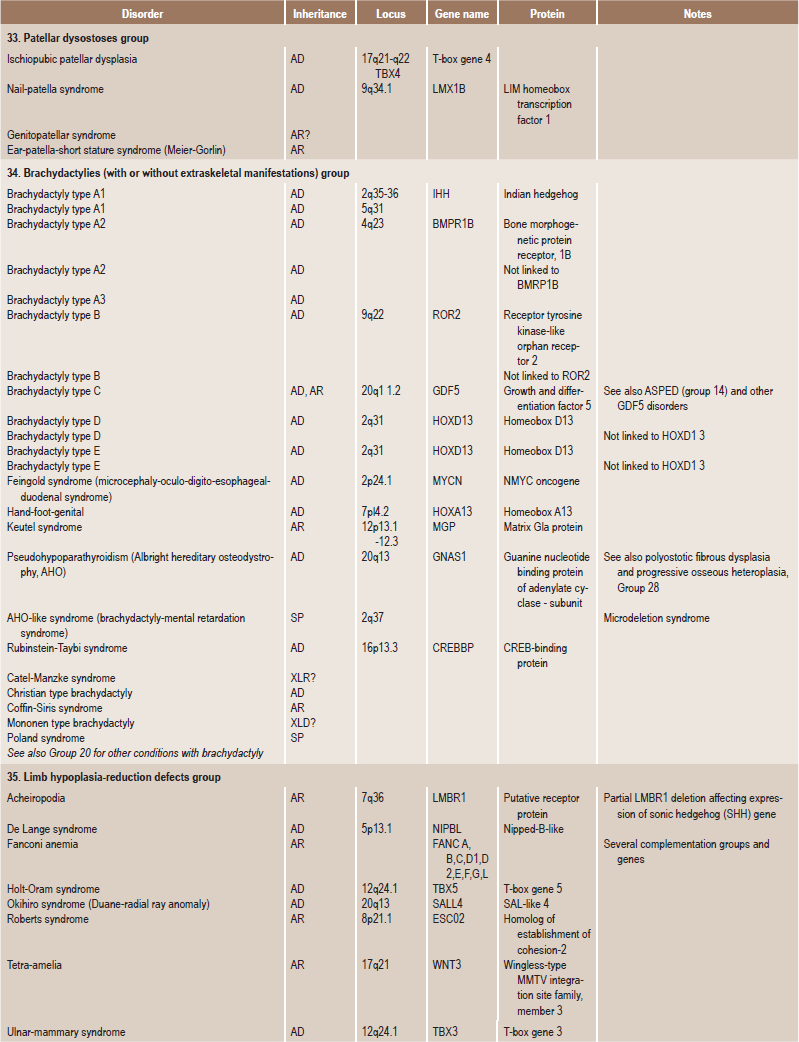
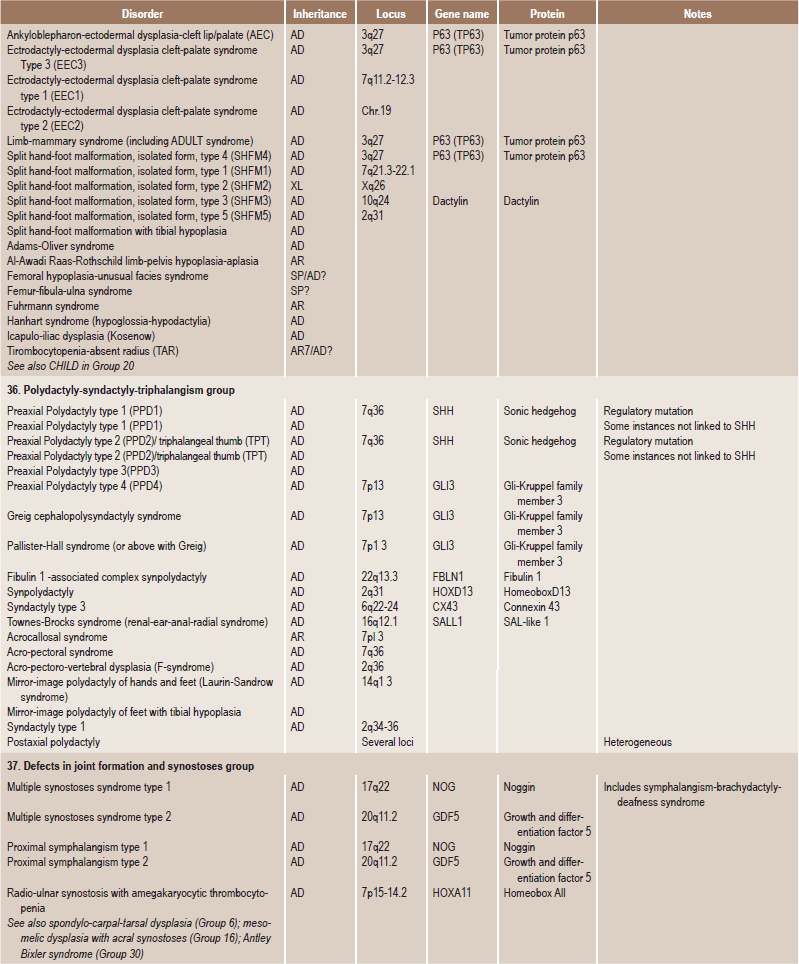
Disorders of Bone-2006, pages 1322–36, Copyright Elsevier, 2007.
Table 17-2
Molecular-pathogenetic classification of genetic disorders of the skeleton
| Gene or protein | Inheritance | Clinical phenotype |
| Group 1: Defects in extracellular structural proteins | ||
| COL1A1, COL1A2 (collagen 1 α1, α2 chains) | AD | Family: Osteogenesis imperfecta |
| COL2A1 (collagen 2 α1 chain) | AD | Family: Achondrogenesis 2, hypochondrogenesis, congenital spondylepiphyseal dysplasia (SEDC), Kniest, Stickler arthro-ophthalmopathy, familial osteoarthritis, other variants |
| COL9A1, COL9A2, COL9A3 (collagen 9 α1, α2, α3 chains) | AD | Multiple epiphyseal dysplasia (MED; two or more variants) |
| COL 10A1 (collagen 10 α1 chain) | AD | Metaphyseal dysplasia Schmid |
| COL 11A1, COL 11A2 (collagen 11 α1, α2 chains) | AR, AD | Oto-spondylo-megaepiphyseal dysplasia (OSMED): Stickler (variant), Marshall syndrome |
| COMP (cartilage oligometic matrix protein) | AD | Pseudoachondroplasia, multiple epiphyseal dysplasia (MED, one |
| MATN3 (matrilin-3) | AD | Multiple epiphyseal dysplasia (MED; one variant) |
| Perlecan | AR | Schwartz-Jampel type 1; dyssegmental dysplasia |
| Group 2: Defects in metabolic pathways (including enzymes, ion channels, and transporters) | ||
| TNSALP (tissue nonspecific alkaline phosphatase) | AR, AD | Hypophosphatasia (several forms) |
| ANKH (pyrophosphate transporter) | AD | Craniometaphyseal dysplasia |
| DTDST/SLC26A2 (diastrophic dysplasia sulfate transporter) | AR | Family: achondrogenesis 1B, atelosteogenesis 2, diastrophic dys-plasia, recessive multiple epiphyseal dysplasia (rMED) |
| PAPSS2, phosphoadenosine-phosphosulfate-synthase 2 | AR | Spondylo-epi-metaphyseal dysplasia Pakistani type |
| TCIRGI, osteoblast proton pump subunit | AR | Severe infantile osteopetrosis |
| CIC-7 (chloride channel 7) | AR | Severe osteopetrosis |
| Carboanhydrase II | AR | Osteopetrosis with intracranial calcifications and renal tubular acidosis |
| Vitamin K-epoxide reductase complex | AR | Chondrodysplasia punctata with vitamin K-dependent coagulation defects |
| MGP (matrix Gla protein) | AR | Keutel syndrome (pulmonary stenosis, brachytelephalangism, cartilage calcifications and short stature) |
| ARSE (arylsulfatase E) | XLR | X-linked chondrodysplasia punctata (CDPXI) |
| 3-β-hydroxysteroid dehydrogenase | XLD | CHILD syndrome |
| 3-β-hydroxysteroid D(8)D(7)-isomerase | XLD | X-linked chondrodysplasia punctata, Conradi-Hunermann type (CDPX2); Child syndrome |
| PEX7 (peroxisomal receptor/importer) | AR | Rhizomelic chondrodysplasia punctata 1 |
| DHAPAT (Dihydroxyacetonphosphate-acyltransferase, peroxisomal enzyme) | AR | Rhizomelic chondrodysplasia punctata 2 |
| Alkyl-dihydroxydiacetonphosphate synthase (AGPS; peroxisomal enzyme) | AR | Rhizomelic chondrodysplasia punctata 3 |
| Group 3: Defects in folding and degradation of macromolecules | ||
| Sedlin (endoplasmic reticulum protein with unknown function) | XR | X-linked spondyloepiphyseal dysplasia (SED-XL) |
| Cathepsin K (lysosomal proteinase) | AR | Pyknodysostosis |
| Lysosomal acid hydrolase and transporters (sulfatase, glycosidase, translocase, etc.) | AR, XLR | Lysosomal storage disease: mucopolysaccharidoses, oligosacchari-doses, glycoproteinoses (several forms) |
| Targeting system of lysosomal enzymes (GlcNAc-1-phosphotransferase) | AR | Mucolipidosis II (I-cell disease), mucolipidosis III |
| MMP2 (matrix metalloproteinase 2) | AR | Torg type osteolysis (nodulosis arthropathy and osteolysis syndrome |
| Group 4: Defects in hormones and signal transduction mechanisms | ||
| 25-α-hydroxycholecalciferol-1-hydroxylase | AR | Vitamin D-dependent rickets type 1 (VDDR1) |
| 1, 25-α-dihydroxy-vitamin D3 receptor | AR | Vitamin D-resistant rickets with end-organ unresponsiveness to vitamin D3 (VDDR 2) |
| CASR (calcium ‘sensor’/receptor) | AD | Neonatal severe hyperparathyroidism with bone disease (if affected fetus in unaffected mother); familial hypocalciuric hypercalcemia |
| PTH/PTHrP receptor | AD (activating mutations) | Metaphyseal dysplasia Jansen |
| AR (inactivating mutation) | Lethal dysplasia Blomstrand | |
| GNAS1 (stimulatory Gs alpha protein of adenylate cyclase) | AD | Pseudohypoparathyroidism (Albright hereditary osteodystrophy and osteodystrophy and several variants) with constitutional haploin-sufficiency mutations; McCune-Albright syndrome with somatic mosaicism for activating mutations |
| PEX proteinase | XL | Hypophosphatemic rickets, X-linked semidominant type (impaired cleav age of FGF23) |
| FGF23, fibroblasts growth factor 23 | AD | Hypophosphatemic rickets, autosomal dominant type (resistance to PEX cleavage) |
| FGFR 1 (fibroblast growth factor receptor 1) | AD | Craniosynostosis syndromes (Pfeiffer, other variants) |
| FGFR 2 | AD | Craniosynostosis syndromes (Apert, Crouzon, Pfeiffer; several variants) |
| FGFR 3 | AD | Thanatophoric dysplasia, achondroplasia, hypochondroplasia, SADDAN: craniosynostosis syndromes (Crouzon with acanthosis nigricans, Muenke nonsyndromic craniosynostosis) |
| ROR-2 (‘orphan receptor tyrosine kinase’) | AR | Robinow syndrome |
| TNFRSF11A (receptor activator of under factor kB; RANK) | AD | Familial expansile osteolysis |
| TGFβ1 | AD | Diaphyseal dysplasia (Camurati-Engelmann) |
| CDMP1 (cartilage-derived morphogenetic protein 1) | AR | Acromesomelic dysplasia Grebe/Hunter-Thompson |
| AD | Brachydactyly type C | |
| Noggin (‘growth factor,’ TGF antegonist) | AD | Multiple synostosis syndrome; synphalangism and hypoacusis syndrome |
| DLL3 (delta-like 3, intercellular signaling) | AR | Spondylocostal dysostosis (one form) |
| IHH (Indian hedgehog signal molecule) | AD | Brachydactyly A1 |
| C7orf2 (orphan receptor) | AR | Acheiropodia |
| SOST (sclerosin; cystine knot secreted protein) | AR | Sclerosteosis, van Buchem disease |
| LRPS (LDL receptor-related protein 5) | AR | Osteoporosis-pseudoglioma syndrome |
| WISP 3 (growth regulator/growth factor) | AR | Progressive pseudorheumatoid dysplasia |
| Group 5: Defects in nuclear proteins and transcription factors | ||
| SOX9 (HMG-type DNA binding protein/ transcription factor) | AD | Compomelic dysplasia |
| GII3 (zinc finger gene) | AD | Greig cephalopolysyndactyly, polydactyly type A and others, Pallister-Hall syndrome |
| TRPS 1 (zine-finger gene) | AD | Tricho-rhino-phalangeal syndrome (types 1–3) |
| HVC (leucine-zipper gene) | AR | Chondroectodermal dysplasia (Ellis-van Creveld) |
| TWIST (helix-loop-helix transcription factor) | AD | Craniosynostosis Saethre-Chotzen |
| P63 (p53 related transcription factor) | AD | EEC syndrome, Hay-Wells syndrome, Limb-mammary syndrome, split hand-split foot malformation (some forms) |
| CBFA-1 (core binding factor A1; runt-type transcription factor) | AD | Cleidocranial dysplasia |
| LXM1B (LIM homeodomain protein) | AD | Nail-patella syndrome |
| DLX3 (distal-less 3 homeobox gene) | AD | Trichodentoosseous syndrome |
| HOXD 13 (homeobox gene) | Ad | Synpolydactyly |
| MSX2 (homeobox gene) | AD (gain of function) | Craniosynostosis, Boston type |
| AD (loss of function) | Parietal foramina | |
| ALX4 (homeobox gene) | AD | Parietal foramina (cranium bifidum) |
| SHOX (short stature-homeobox gene) | Pseudo-autosomal | Leri-Weill dyschondrosteosis, idiopathic short stature |
| TBX3 (T-box 3, transcription factor) | AD | Ulnar-mammary syndrome |
| TBX5 (T-box 5, transcription factor) | AD | Holt-Oram syndrome |
| EIF2AK3 (transcription initiation factor kinase) | AR | Wolcott-Rallison syndrome (neonatal diabetes mellitus and spondy-loepiphyseal dysplasia) |
| NEMO (NFkB essential modulator; kinase activity) | LX | Osteopetrosis, lymphedema, ectodermal dysplasia and immunode-ficiency (OLEDAID) |
| Group 6: Defects in oncogenes and tumor suppressor genes | ||
| EXT1, EXT2 (exostosin-1, exostosin-2; heparan-sulfate polymerases) | AD | Multiple exostoses syndrome type 1, type 2 |
| SH3BP2 (a-Abl-binding protein) | AD | Cherubism |
| Group 7: Defects in RNA and DNA processing and metabolism | ||
| RNAse MRP-RNA component | AR | Cartilage-hair-hypoplasia |
| ADA (adenosine deaminase) | AR | Severe combined immunodeficiency (ACID) with (facultative) metaphyseal changes |
Definitions
The clinical evaluation should start with a complete medical history that includes previous milestones of growth. Since skeletal dysplasias may become apparent at various ages, study of growth points since birth may help to narrow the differential diagnosis. The family history should include information about other affected family members and possible consanguinity. Parents should be examined for evidence of disproportionate stature or other evidence of a skeletal dysplasia. Physical examination should focus on anthropometric measurements. The osteochondrodysplasias are generalized disorders of the skeleton, which usually result in disproportionate short stature. A disproportionate body habitus may not be readily appreciated unless anthropometric measurements (i.e. arm span, upper to lower segment ratio, etc.) are carefully obtained. This assessment may help to determine if the disproportionate shortening affects primarily the trunk or the limbs [the proximal (rhizomelic), middle (mesomelic) or distal segment (acromelic)].
Group I: Defects in Extracellular Structural Proteins (Based on Molecular Pathogenetic Classification of Genetic Disorders of Skeleton)
Osteogenesis Imperfecta (‘Brittle bones’, fragilitas ossium, osteopsathyrosis, Lobstein’s disease)
Osteogenesis imperfecta (OI) is a serious disease, the molecular pathogenesis of which is being elucidated and it bears a superficial relatedness to dentinogenesis imperfecta (refer Chapter 1, section on dentinogenesis imperfecta), a milder condition affecting mesodermal tissues. It is a condition resulting from abnormality in the type I collagen, which most commonly manifests as fragility of bones. Although osteogenesis imperfecta is generally recognized as representing a hereditary autosomal dominant characteristic, autosomal recessive and nonhereditary types also occur.
Four types of osteogenesis imperfecta exist (Table 17-3), based on the classifications of Sillence et al (1979).
Table 17-3
Clinical types of osteogenesis imperfecta
Osteogenesis imperfecta, type I
Osteogenesis imperfecta tarda
Osteogenesis imperfecta with blue sclerae
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta congenita; type II
Osteogenesis imperfecta congenita, neonatal lethal
Vrolik type of osteogenesis imperfecta
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta, progressively deforming, with normal sclerae: type III
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta, type IV
Osteogenesis imperfecta with normal sclerae
Gene map locus 17q21.31–q22
Clinical Features
The chief clinical characteristic of osteogenesis imperfecta is the extreme fragility and porosity of the bones, with an attendant proneness to fracture. The fractures heal readily, but the new bone is of a similar imperfect quality.
Physical features can vary depending on the type. It forms the basis for Sillence classification.
Histologic Findings
The bones in patients with osteogenesis imperfecta exhibit thin cortices, sometimes being composed of immature spongy bone, while the trabeculae of the cancellous bone are delicate and often show microfractures (Fig. 17-1). Osteoblastic activity appears retarded and imperfect, and for this reason the thickness of the long bones is deficient. The basic defect appears to lie in the organic matrix with failure of fetal collagen to be transformed into mature collagen. Qualitative defects (abnormal collagen I molecule) and quantitative defects (decrease in production of normal collagen I molecules) both exist. There is some evidence that the progressive intermolecular cross-linkage of adjacent collagen molecules, which is an essential characteristic of normal collagen maturation, is defective in this disease. Calcification proceeds normally. Defective microvascular system and decreased collagen fibril diameter have also been observed. The length of the long bones is usually normal unless multiple fractures have caused undue shortening.
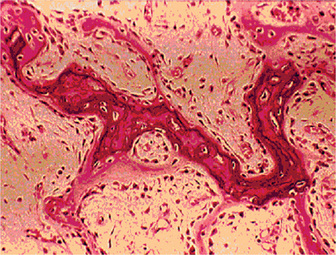
Figure 17-1 Osteogenesis imperfecta (OI).
The typical microscopic changes of OI can be seen in a section of a long bone of a severely affected child. The bone cortex is thin and porous. The bone trabeculae are thin, delicate, and widely separated. Many osteoblasts and osteocytes are present, but the formation and organization of osteoid is deficient. There is less bone tissue than normal and most of it is woven or nonlamellar bone with collagen fibers of small size and random distribution. The woven bone has an increase in basophilic ground substance (shown by blue staining in H and E sections). (Courtesy of Dr Robert C Mellors)
Treatment and Prognosis
There is no known treatment for osteogenesis imperfecta. No medical therapy is involved, other than the treatment of infections when they occur. The prognosis varies from relatively good to very poor. In type IA, life expectancy is similar to that of general population; type II, most patients die within the first year of life. A slight decrease in life expectancy has been observed in the other types.
Marfan Syndrome (Marfan-Achard syndrome, arachnodactyly)
Clinical Features
The estimated incidence of Marfan syndrome ranges from 1 in 5,000 to 1 in 10,000 births which includes stillbirths. The wide variation in the sites of mutations noticed in the fibrillin gene causes the varied phenotypic manifestations of this syndrome. Several other diseases present similar to Marfan syndrome, making it exceedingly difficult to determine the exact incidence. The skeleton typically displays multiple deformities including arachnodactyly, dolichostenomelia (i.e. long limbs relative to trunk length), and thoracolumbar scoliosis. The shape of the skull and face is characteristically long and narrow, and commonly suggests the diagnosis of the disease (Figs. 17-2, 17-3). Other features of the disease include hyperextensibility of joints with habitual dislocations, kyphosis and flat feet. In the cardiovascular system, aortic dilation, aortic regurgitation, and aneurysms are the most worrisome clinical findings. Mitral valve prolapse requiring valve replacement can occur as well. Ocular findings include myopia, cataracts, retinal detachment, and superior dislocation of the lens.

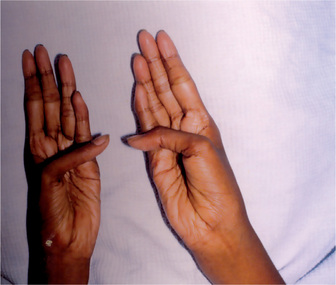
Oral Manifestations
According to Baden and Spirgi, who have reviewed the oral manifestations of this disease, a high, arched palatal vault is very prevalent and may be a constant finding. Bifid uvula is also reported as well as malocclusion. In addition, multiple odontogenic cysts of the maxilla and mandible have occasionally been reported, most recently by Oatis and his coworkers. One additional finding sometimes present, is temporomandibular dysarthrosis (Fig. 17-4).
Achondrogenesis
Clinical Features
In achondrogenesis type I, the craniofacial features include a disproportionately large head, soft skull, sloping forehead, convex facial plane, flat nasal bridge, occasionally associated with a deep horizontal groove, small nose, often with anteverted nostrils, long philtrum, retrognathia, increased distance between lower lip and lower edge of chin and double chin appearance (often). In achondrogenesis type II, the features seen are a disproportionately large head, large and prominent forehead, flat facial plane, flat nasal bridge, small nose with severely anteverted nostrils, normal philtrum (often), micrognathia (Fig. 17-5). The differential diagnoses include achondroplasia, hypophosphatasia, osteogenesis imperfecta and thanatophoric dysplasia.
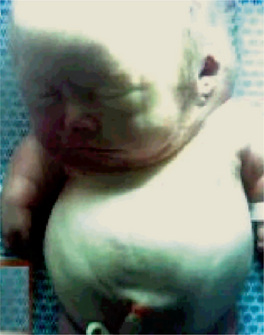
Figure 17-5 An infant with achondrogenesis type II.
Note the disproportionately large head, large and prominent forehead, flat facial plane, flat nasal bridge, small nose with severely anteverted nostrils, micrognathia, extremely short neck, short and flared thorax, protuberant abdomen, and extremely short upper extremities.
Radiographic Features
The radiographic features may vary, and no single feature is consistently noticed. Distinction between type IA and type IB on radiographs is not always possible. Degree of ossification is age dependent, and caution is needed when comparing radiographs at different gestational ages.
Hypophosphatasia
Clinical Features
The only physical finding in the odontohypophosphatasic form is the premature loss of teeth.
Radiographic Features
The childhood form is characterized by rachitic deformities. Upon radiologic examination of the metaphysis, evidence of radiolucent projections from the epiphyseal plate into the metaphysis is present. This is not found in other types of rickets. Radiographic findings are normal for patients with odontohypophosphatasia. Dental radiographs generally reveal hypocalcification of teeth and the presence of large pulp chambers, as well as alveolar bone loss; however these findings have not been consistently reported.
Osteopetrosis (Marble bone disease, Albers-Schönberg disease, osteosclerosis fragilis generalisata)
Oral Manifestations
The jaws are involved in the same manner as the other bones in the body, and the oral manifestations have been reviewed by Kaslick and Brustein. However, a clear distinction has usually not been made as to the type of the disease present, benign or malignant. The medullary spaces of the jaws are remarkably reduced in both dominant and recessive osteopetrosis so that there is a marked predilection for the development of osteomyelitis should infection gain entrance to the bone. This is a complication of dental extraction which has been reported frequently and discussed by Dyson. Similar findings were noted by Bjorvatn and his associates in four children with the malignant form of the disease. They stressed the necessity of administering large doses of antibiotics to control the recurring infection, which even then did not prevent the progressive osseous destruction. Fracture of the jaw during tooth extraction, even when the extraction is performed without undue force, may also occur because of the fragility of the bone. It has been reported that the teeth are of defective quality, enamel hypoplasia, microscopic dentinal defects and arrested root development all having been described. However, this may not be true in the benign dominant form of the disease. It is also reported that the teeth are especially prone to dental caries. Since dental findings have been recorded in so few cases, this observation is difficult to evaluate. An additional rather constant finding is retardation of tooth eruption due to the sclerosis of bone.
Radiographic Features
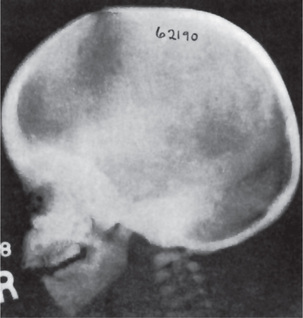
Radiographic features are usually diagnostic. Because the disease is a heterogeneous group of disorders, the findings vary depending on the subtype. Patients usually have generalized osteosclerosis. Bones may be uniformly sclerotic, but alternating sclerotic and lucent bands may be noted near the ends of long bones (Fig. 17-6). The bones might appear club like or show an appearance of a bone within bone (endobone). The entire skull is thickened and dense, especially at the base. Sinuses are small and underpneumatized. Vertebrae are extremely radiodense. They may show alternating bands, known as the ‘rugger-jersey’ sign. Radiographs may show evidence of fractures or osteomyelitis. When the jaws are affected, the density of the bone may be such that the roots of the teeth are nearly invisible on the dental radiograph.
Histologic Features
Bone biopsy is not essential for diagnosis because radiographs are usually diagnostic. Osteopetrosis is characterized by the endosteal production of bone with an apparent concomitant lack of physiologic bone resorption (Fig. 17-7). Osteoblasts are prominent, but osteoclasts are seldom found in significant numbers in tissue sections. The predominance of bone formation over resorption typically leads to the persistence of cartilaginous cores of bony trabeculae long after their replacement should have occurred in endochondral bones. The trabeculae themselves are disorderly in arrangement, and the marrow tissue present is usually fibrous.
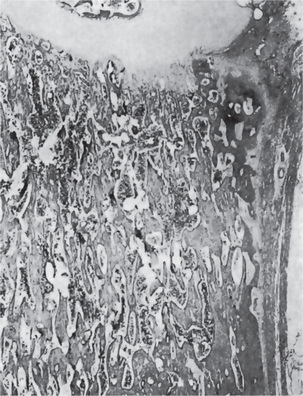
Figure 17-7 Osteopetrosis.
A photomicrograph of a long bone showing replacement of the marrow by endosteal bone. (Courtesy of Dr Frank Vellios)
It has been reported by Johnston and his associates; however, that adult patients with benign osteopetrosis do not appear to have a deficiency in osteoclastic activity but rather an abnormality in the type and structure of bone. They found osteoblastic and osteoclastic activity with prominent remodeling of bone. However, by polarized light, the bone was found to be markedly deficient in collagen matrix fibrils and these seldom crossed from one osteon to another. This deficiency of fibrils could account for the tendency for fracture in these patients.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


